Regulatory Preclinical Strategy — Expert Guide
IND Submission Preclinical Requirements: A Practical Guide to a Regulatory-Ready Nonclinical Package
Translating a promising molecule or medical device from the bench to first-in-human (FIH) trials is one of the most demanding transitions in drug development. The IND submission preclinical requirements define the scientific and regulatory bar a sponsor must meet to demonstrate that initiating clinical trials is reasonably safe. This guide walks biotech teams, researchers, and regulatory affairs professionals through what a compliant regulatory preclinical package looks like, how to interpret FDA IND submission checklist expectations, and how to structure robust preclinical data for IND filings without unnecessary delays.
The aim is not a complete safety profile suitable for marketing authorization, but rather “sufficient safety to proceed” into early clinical exposure. With three decades of large-animal preclinical experience supporting medical device and pharmaceutical sponsors, Biotech Farm has seen first-hand how the right study design, documentation discipline, and scientific escort can shorten the path between discovery and clinic.
🔎 Expert Insight: What Regulators Are Really Looking For
Regulators do not expect perfection — they expect scientific coherence. A nonclinical package that connects mechanism of action to observed toxicity, and observed toxicity to clinical monitoring, signals the kind of translational maturity that accelerates IND review. Programs that present studies as disconnected documents, rather than an integrated safety narrative, routinely attract additional information requests that add months to the clock.
What Are the Core Preclinical Requirements for an IND Submission?
To file an Investigational New Drug application, sponsors must compile a nonclinical data package that supports the reasonable safety of initiating human trials. The package typically integrates pharmacology (primary and secondary), toxicology, safety pharmacology, and ADME/PK characterization, all tailored to the product’s modality, route of administration, and clinical plan.
Per 21 CFR § 312.23, the IND must contain a dedicated nonclinical section summarizing study designs, results, safety conclusions, and a GLP compliance statement for each study. The emphasis is on demonstrating that the proposed clinical protocol is supported by an adequate safety margin and that monitoring strategies address identified risks.
How Do IND-Enabling Studies Differ From Early Discovery Research?
Early discovery research is exploratory: it generates hypotheses, supports candidate selection, and may rely on partial endpoints, non-validated assays, and non-GLP conditions. IND-enabling studies, in contrast, are deliberately structured to support FIH safety. They include repeat-dose toxicology in relevant species, safety pharmacology core battery, genotoxicity (for small molecules), and ADME/PK characterization.
The two stages also differ in documentation rigor. IND-enabling studies require pre-approved protocols, traceable raw data, audited reports, and clearly defined endpoints, while discovery work is typically lab-notebook driven. Sponsors who blur this distinction often face regulatory questions about data integrity at the worst possible moment.
⚠️ Common Mistake to Avoid
Treating discovery-phase, non-GLP data as IND-enabling evidence is one of the most frequent causes of clinical hold. Regulators review the GLP compliance statement for every pivotal safety study — if it is missing or inadequate, the submission stalls.
The Building Blocks of the Nonclinical Package
Each component of the nonclinical package answers a distinct regulatory question. Understanding the role of each block helps sponsors plan studies that integrate, rather than duplicate, evidence.
| Block | Question Addressed | Typical Studies |
|---|---|---|
| Primary Pharmacology | Does it work? | Efficacy in disease-relevant models, dose-response, biomarkers |
| Secondary Pharmacology | What else might it touch? | Receptor/enzyme panels, off-target screens |
| Safety Pharmacology | What is the acute risk to vital systems? | Cardiovascular, respiratory, CNS (ICH S7A core battery) |
| Toxicology | Where does it cause harm? | Repeat-dose tox, genotoxicity, local tolerance |
| ADME/PK | What is the exposure? | Absorption, distribution, metabolism, excretion, TK |
Requirements Roadmap by Development Stage
ICH M3(R2) introduces a “graded approach”: some studies are mandatory before FIH (acute and subchronic toxicology, safety pharmacology core battery, basic PK), while reproductive toxicity, carcinogenicity, and chronic toxicology can be staged across later phases. Mapping required studies against the planned clinical exposure duration is the most efficient way to avoid over- or under-investing in nonclinical work.
Inside the Pharmacology and Toxicology Section

The Pharm/Tox section must include an integrated summary of all relevant nonclinical studies, accompanied by full reports, tabulated data, and appendices. Reviewers expect to see the rationale for model selection, dosages tested, achieved exposures, observed findings, NOAEL/LOAEL determinations, and explicit conclusions for the proposed clinical program.
A common mistake is presenting studies as standalone documents. Regulators read the section as a narrative: mechanism of action leads to expected pharmacological effects, which lead to anticipated and observed toxicities, which lead to clinical monitoring choices. When that thread is missing, additional information requests are nearly inevitable.
Telling a Coherent Regulatory Story
Connect the dots explicitly. If primary pharmacology suggests a cardiovascular mechanism, the safety pharmacology and toxicology data should address cardiac endpoints, and the clinical protocol should include corresponding monitoring (ECG, blood pressure, biomarkers). This integration signals scientific maturity and reduces reviewer uncertainty.
“A nonclinical package should read like a scientific argument, not a filing cabinet. Each study adds a chapter to the same story — and the conclusion must justify initiating human exposure.”
— Adir Koreh, CEO, Biotech Farm Ltd.
Is GLP Compliance Required for Every Preclinical Study?
Not every study must be GLP-compliant. Definitive safety studies supporting the IND — primarily pivotal toxicology and safety pharmacology — are expected to follow 21 CFR Part 58. Efficacy pharmacology studies may be non-GLP if methods, results, and quality controls are well documented. GLP exists to ensure traceability, data integrity, and reproducibility, not to dictate scientific design.
Handling GLP Deviations Without Jeopardizing the Submission
If a study deviated from GLP, disclose it transparently: explain the reason, assess the impact on data interpretation, and describe mitigation steps such as independent QA review or supplementary data. Hidden deviations discovered during inspection are far more damaging than disclosed and justified ones.
✅ GLP Required (Pivotal Studies)
- Repeat-dose toxicology studies
- Safety pharmacology core battery
- Genotoxicity battery (small molecules)
- Local tolerance studies
???? Non-GLP Acceptable (Exploratory)
- Primary pharmacology / efficacy models
- Range-finding and dose-ranging studies
- Secondary pharmacology screens
- Mechanism-of-action investigations
Pharmacology Studies Expected Before an IND
Regulatory bodies expect primary pharmacology that demonstrates activity and mechanism in models relevant to the proposed indication, including dose-response data and biomarker correlation when feasible. Secondary pharmacology screens broader receptor and enzyme panels to flag off-target liabilities that could translate into clinical adverse events.
For complex products such as implantable devices, combination therapies, or interventional cardiology technologies, large-animal models often provide the most translatable evidence. Biotech Farm’s surgical infrastructure — including fluoroscopy, high-definition ultrasound, echocardiography, and 4K laparoscopic platforms — is configured to deliver pharmacology and proof-of-concept data under conditions that mirror clinical practice.
Why Safety Pharmacology Is Critical Before First-in-Human
Safety pharmacology investigates undesirable effects on vital organ systems — cardiovascular, respiratory, and central nervous — to mitigate the risk of acute adverse events at trial initiation. The ICH S7A core battery is the baseline expectation, and findings directly inform initial dose selection, escalation rates, and clinical monitoring such as ECG, respiration tracking, and neurological assessment.
Core Battery vs. Follow-Up Studies
The core battery (cardiovascular, respiratory, CNS) is sufficient when no signals emerge and the product class lacks known liabilities. When signals appear, or when class effects (e.g., QT prolongation per ICH S7B) are anticipated, follow-up studies become necessary. Cardiac repolarization assessment frequently combines hERG in vitro testing with in vivo telemetry in a non-rodent species.
✅ Best Practice: Integrated Cardiovascular Assessment
For cardiovascular-active compounds or devices, combine hERG channel binding in vitro with in vivo telemetry (hemodynamics + ECG) in a swine or canine model. This dual-layer approach satisfies ICH S7A/S7B simultaneously and provides translatable data that directly informs clinical ECG monitoring intervals.
Toxicology Studies Required for an IND Submission
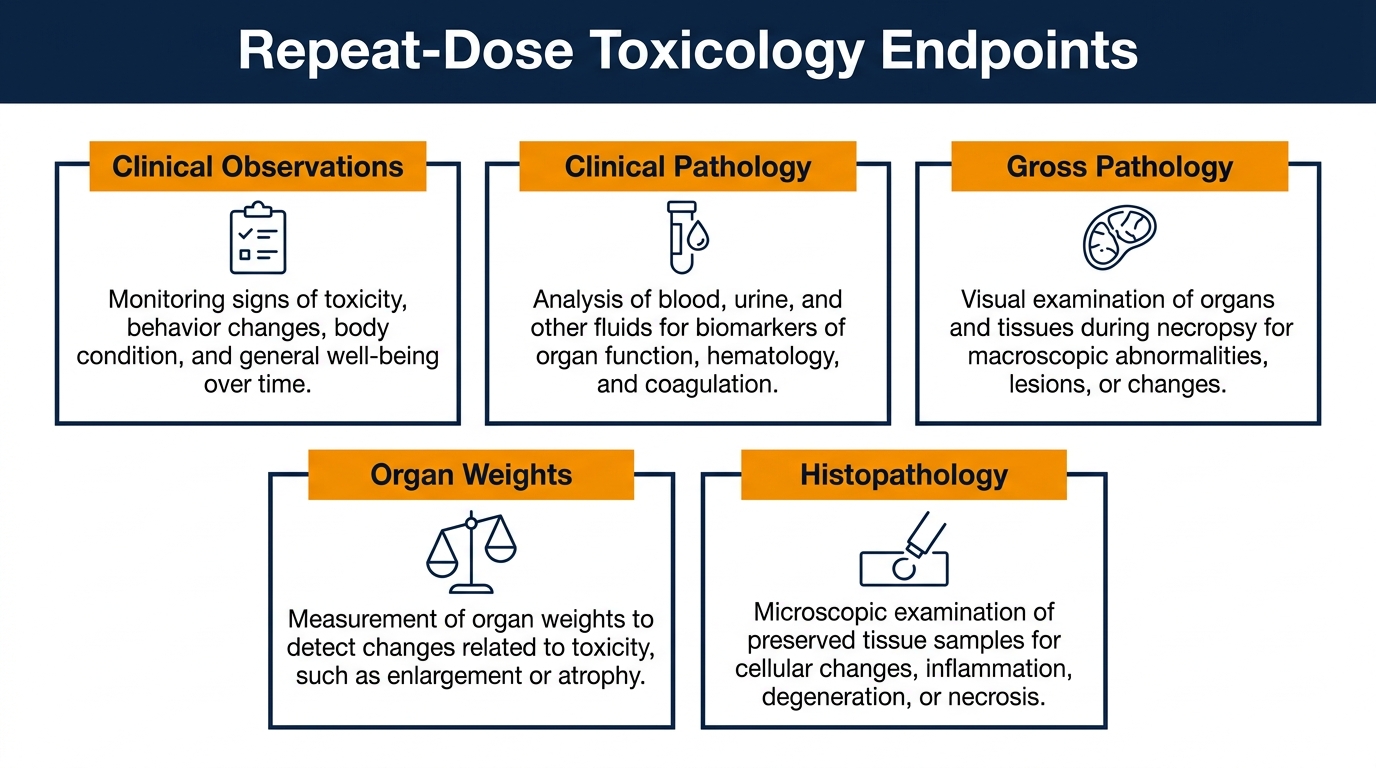
Repeat-dose toxicology studies must support the planned clinical exposure duration and route of administration. Endpoints include clinical observations, body weight, food consumption, clinical pathology (hematology, chemistry, urinalysis), gross pathology, organ weights, and histopathology. The output identifies target organs, establishes safety margins, and guides clinical monitoring strategies.
Single-Dose vs. Repeat-Dose Toxicology
Single-dose studies characterize acute toxicity and inform initial dose escalation in FIH trials. Repeat-dose studies — typically 14 to 28 days for short clinical exposure or up to 3–6 months for longer programs — characterize cumulative toxicity, recovery, and reversibility. Duration must equal or exceed the proposed clinical exposure.
How Many Species Are Needed for Toxicology?
The default expectation for small molecules is two species: a rodent (typically rat) and a non-rodent (often dog, minipig, or non-human primate). Species selection must be justified on biological and pharmacological relevance, target expression, metabolic similarity to humans, and exposure achievability — not laboratory convenience. Pig models are particularly valuable for cardiovascular, dermal, and gastrointestinal programs given anatomical and physiological similarity to humans.
Justifying Single-Species Toxicology
For biologics covered by ICH S6(R1), only one pharmacologically relevant species may be acceptable when binding or activity is demonstrated in only one non-human species. The justification file must include cross-species target homology data, in vitro binding studies, and a discussion of why a second species would not add scientific value.
CASE STUDY: Swine Model for Cardiovascular Device IND
Challenge: Demonstrating device safety in a single relevant species
A cardiovascular device sponsor required IND-enabling toxicology for an interventional implant with no rodent-relevant anatomy. Biotech Farm designed a swine-only program justified by coronary artery size, cardiac physiology, and coagulation profile analogous to humans. ICH S6(R1) single-species rationale was formally documented.
Outcome: FDA accepted the single-species justification at pre-IND meeting. The IND submission cleared without clinical hold. FIH trial initiated within 14 months of program start.
Determining the First-in-Human Dose From Preclinical Data
The FIH dose is derived from NOAEL data in the most sensitive relevant species, converted to a Human Equivalent Dose (HED) using allometric scaling, and adjusted by a safety factor (typically 10-fold, but higher for steep dose-response curves, irreversible toxicity, or novel mechanisms). The Maximum Recommended Starting Dose (MRSD) is then compared against pharmacologically active dose estimates to confirm an informative yet conservative starting point.
Common Mistakes in FIH Calculation
Frequent pitfalls include ignoring active metabolites, applying scaling factors inappropriate for the route of administration, overlooking nonlinear PK at higher doses, and failing to address bioavailability differences between animal and human formulations. Each of these can lead to either an unsafe starting dose or one so low that the trial loses scientific value.
???? FIH Dose Calculation Framework
- Step 1: Identify NOAEL from most sensitive species
- Step 2: Convert to HED using body surface area scaling
- Step 3: Apply safety factor (typically 10×, higher for novel mechanisms)
- Step 4: Compare MRSD against pharmacologically active dose
- Step 5: Justify starting dose in IND clinical protocol section
How Much ADME/PK Data Is “Sufficient”?
A working understanding of absorption, distribution, metabolism, and excretion — together with toxicokinetic exposure data from pivotal toxicology studies — is typically required. PK data enables interspecies exposure comparison, supports dose selection, identifies metabolites of concern, and informs drug-drug interaction risk. For biologics, immunogenicity assessment in toxicology species is also expected.
Common Pitfalls in Nonclinical IND Submissions
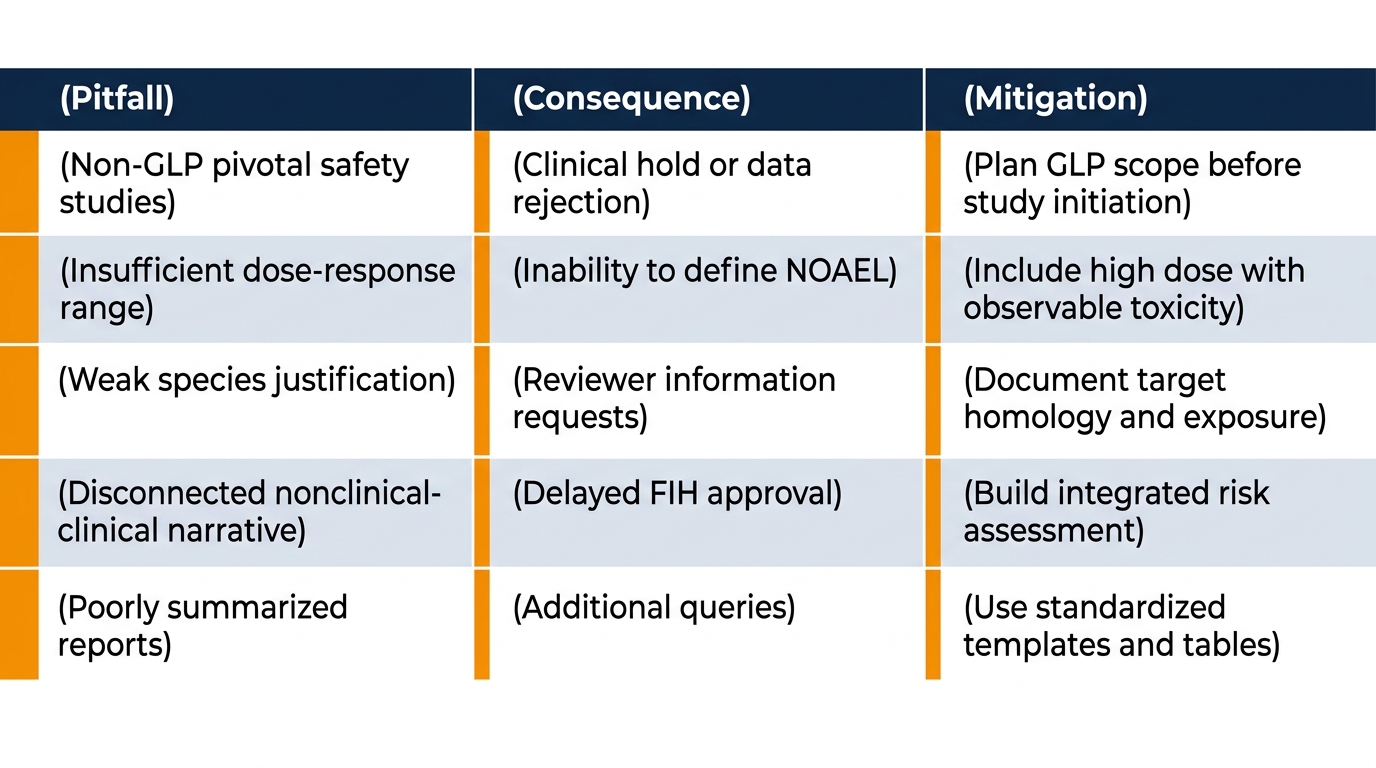
| Pitfall | Consequence | Mitigation |
|---|---|---|
| Non-GLP pivotal safety studies | Clinical hold or data rejection | Plan GLP scope before study initiation |
| Insufficient dose-response range | Inability to define NOAEL/MTD | Include high dose with observable toxicity |
| Weak species justification | Reviewer information requests | Document target homology and exposure |
| Disconnected nonclinical-clinical narrative | Delayed FIH approval | Build integrated risk assessment |
| Poorly summarized reports | Reviewer fatigue, additional queries | Use standardized templates and tables |
How a Pre-IND Meeting Strengthens Your Nonclinical Strategy
A pre-IND meeting is an opportunity to align with the FDA on study design, species choice, GLP scope, and clinical protocol elements before significant investment. Sponsors submit a briefing document with specific questions, and FDA feedback either confirms the planned approach or flags gaps while there is still time to address them. For complex products — gene therapies, combination devices, novel modalities — this dialogue can save months of rework.
For an in-depth view of how IND-enabling work translates into a structured filing strategy, see Biotech IND-enabling preclinical studies and filing.
Building Your IND Submission Preclinical Requirements Checklist
A practical checklist anchors the team during the months leading to submission. It should map every required study to its GLP status, completion date, report number, and integration into the IND nonclinical summary. Endpoints to track include pharmacology models, toxicology duration and species, safety pharmacology core battery, genotoxicity battery, ADME/PK characterization, and formulation bridging if the clinical formulation differs from the toxicology formulation.
External references such as Preclinical Data for IND Applications can complement internal templates and help cross-check completeness. An additional operational perspective is available at Pre-Clinical Checklist for IND Submissions.
???? Essential IND Nonclinical Checklist Items
- Primary pharmacology efficacy models with dose-response data
- GLP repeat-dose toxicology in 2 relevant species (or justified 1)
- Safety pharmacology core battery (ICH S7A): CV, respiratory, CNS
- Genotoxicity battery (small molecules): Ames, chromosomal aberration, in vivo
- ADME/PK characterization with toxicokinetic sampling
- GLP compliance statements for all pivotal studies
- NOAEL/LOAEL identification and MRSD calculation
- Formulation bridging data if clinical and tox formulations differ
Mapping Sponsor Needs to Preclinical Capabilities
| Sponsor Need | What a Specialized Large-Animal Facility Provides |
|---|---|
| GLP-grade safety data on a medical device | Validated procedures, audit-ready documentation, and trained surgical teams |
| Translational pharmacology in cardiology or orthopedics | Pig, sheep, and goat models with imaging and surgical capabilities |
| Project agility under tight timelines | Integrated scientific escort, in-house imaging, and a single point of accountability |
| Ethical and regulatory transparency | 3R-aligned protocols, well-documented procedures, and IACUC oversight |
| Adaptation to novel modalities | Tailored study designs developed with the sponsor’s regulatory plan in mind |
Navigating the FDA IND Submission Checklist
The structural backbone of an IND is defined in 21 CFR § 312.23: cover sheet (Form FDA 1571), table of contents, introductory statement, general investigational plan, investigator brochure, clinical protocol, chemistry/manufacturing/controls (CMC), pharmacology and toxicology information, previous human experience, and additional information. Each nonclinical study report must carry a clear GLP statement.
For a complementary perspective, the IND Application Checklist outlines submission readiness from a reviewer’s standpoint.
Special Considerations for Biologics and Gene Therapies
Biologics often require species-specific design considerations because activity may be confined to one or two species expressing the target. Immunogenicity assessment, anti-drug antibody monitoring, and neutralizing activity become integral to interpreting toxicology results. Gene therapies introduce additional questions: vector biodistribution, integration risk, germline transmission, and shedding all require dedicated nonclinical studies that go beyond conventional small-molecule frameworks.
A reference dataset structure for these programs is illustrated in this Preclinical Data Package for IND Submission.
The Role of Translational Science in IND Preparation
Translational science bridges nonclinical findings and clinical outcomes. It informs biomarker selection, exposure-response modeling, and the design of FIH studies that maximize learning per patient exposed. When the preclinical team and the clinical team share a translational framework, dose escalation rules, stopping criteria, and safety monitoring become scientifically grounded rather than arbitrarily conservative.
“The best preclinical package is not the biggest one — it is the one that answers every question the reviewer will have before they ask it. That requires deep translational thinking, not just experimental execution.”
— Adir Koreh, CEO & Founder, Biotech Farm Ltd.
Ensuring Data Quality and Integrity Throughout
Quality is not added at the end; it is engineered into the program from protocol design through final report. GLP studies require pre-approved protocols, validated equipment, trained personnel, contemporaneous raw data, and independent QA review. Beyond GLP, data integrity principles (ALCOA+: attributable, legible, contemporaneous, original, accurate, complete, consistent, enduring, available) apply to every record that may support a regulatory submission.
ALCOA+ Principles for IND Data
- Attributable — every record traced to its author
- Legible — readable throughout its lifetime
- Contemporaneous — recorded at time of activity
- Original — first-capture record retained
- Accurate — free from errors and omissions
- + Complete, Consistent, Enduring, Available
Frequently Asked Questions
Ready to Build a Preclinical Package That Stands Up to Regulatory Scrutiny?
Whether your program involves a novel medical device, a biologic, or a complex combination product, the quality of your nonclinical data will define how smoothly your IND moves through review. Have you mapped every IND submission preclinical requirement to a study, a timeline, and a GLP plan?

This article is intended for informational purposes for biotech, pharmaceutical, and medical device professionals. For regulatory guidance specific to your program, consult qualified regulatory affairs counsel and refer to current FDA and ICH guidelines.




