Repeat Dose Toxicity Study in Animals: A Practical Definition
A repeat dose toxicity study in animals is a structured preclinical safety assessment in which a test article — a pharmaceutical, medical device extractable, biocide, pesticide, or industrial chemical — is administered to laboratory animals on a recurring schedule across a defined window, commonly 28 or 90 days. The study is engineered to expose target organs, characterize dose-response behavior, and anchor regulatory reference points such as the No Observed Adverse Effect Level (NOAEL). Unlike a single-shot acute test, repeated administration captures cumulative pharmacodynamics, adaptive responses, and emerging histopathological signals. The data feeds directly into quantitative risk assessment and IND/CE-mark dossiers, and supports translational decisions about first-in-human dosing. At BIOTECH FARM, more than 30 years of preclinical R&D experience inform tailored protocols that match scientific objectives to regulatory expectations from the earliest design stage.
Without repeat-dose toxicity data, sponsors risk discovering target-organ toxicity only in patients — the most expensive and least ethical place to find it. A well-designed multi-dose study is not a regulatory hurdle; it is the strategic foundation of every defensible clinical development program.
Beyond Acute Testing: Why Repeated Exposure Reveals What a Single Dose Cannot
Acute studies report what happens when an organism encounters a substance once. Real-world exposure — whether a chronic medication, an implanted device leachable, or occupational chemical contact — rarely behaves that way. Repeated dosing surfaces effects that a single administration simply cannot generate: bioaccumulation in lipid-rich tissues, induction or suppression of metabolic enzymes, progressive renal or hepatic stress, immunological sensitization, and delayed organ-specific lesions.
Regulatory frameworks therefore separate acute from subchronic and chronic categories on purpose, since each duration interrogates a different biological question. Without a repeat-dose phase, a sponsor risks discovering target-organ toxicity only in patients — the most expensive and least ethical place to find it.
Comparing 28-Day, 90-Day, and Chronic Toxicity Testing
Selection between durations is rarely arbitrary. A 28-day study is a fast, focused screen — frequently used to bracket dose ranges, support early formulation decisions, or satisfy abbreviated regulatory pathways. A 90-day subchronic design is the workhorse for systemic safety profiling and feeds dose selection for longer programs. Chronic studies extend exposure over six to twelve months or beyond, probing late-onset pathology and cumulative carcinogenic potential.
| Study Type | Typical Duration | Primary Purpose | Common Use Case |
|---|---|---|---|
| Short-term repeat dose | 14–28 days | Screening, dose-range finding | Early candidate triage |
| Subchronic | 90 days | Target organ ID, NOAEL | Pivotal IND-enabling package |
| Chronic | 6–12 months | Long-term systemic safety | Marketed-drug or lifetime exposure |
What a Subchronic Toxicity Study Actually Delivers
A subchronic toxicity study, generally executed over a 90-day continuous dosing window, is designed to expose systemic effects that escape shorter screens. It identifies target organs, characterizes the gradient of dose-response, and produces the NOAEL value that underpins downstream chronic, reproductive, or carcinogenicity protocols. Subchronic data also flag where additional mechanistic work is warranted — for example, when histopathology reveals hepatocellular hypertrophy that may or may not represent adverse pathology.
Because the 90-day endpoint must be defensible across multiple jurisdictions, study integrity hinges on tightly controlled environmental conditions, validated analytical methods, and transparent documentation. A scientifically supportive facility with senior surgeons, dedicated study directors, and well-documented procedures protects both the animals and the regulatory value of the dataset.
Position Within a Broader Development Program
Subchronic results rarely stand alone. They function as the bridge between exploratory pharmacology and pivotal long-term safety studies, and they often determine whether a candidate progresses, pivots formulation, or is reformulated entirely. Sponsors who treat the 90-day study as a regulatory checkbox tend to lose time later; sponsors who treat it as a strategic decision point save it.
Selecting Test Species: Rodent, Non-Rodent, or Both

Species selection is governed by biological and pharmacological relevance, regulatory expectations, and the practical capacity to measure exposure and response reliably. Most pharmaceutical programs require both a rodent and a non-rodent species because each offers complementary translational signal. Rodents deliver statistical power, established historical control data, and efficient throughput. Non-rodent models — pigs, rabbits, sheep, or goats depending on the indication — provide anatomical and physiological proximity to humans for cardiovascular, ophthalmologic, orthopedic, and device-related questions.
BIOTECH FARM‘s large animal facility supports porcine, ovine, caprine, and rabbit models, with humanoid organ scale that suits cardiology, wound healing, respiratory, metabolic, and ophthalmology platforms.
When a Single Species May Be Justified
Single-species studies are accepted only when supported by strong scientific rationale — for instance, when receptor expression, pharmacology, or target biology is meaningful in only one species. Such deviations require pre-submission alignment with the relevant authority and rigorous documentation of the underlying justification.
How Many Animals Are Required
Group sizing is dictated by guideline, species, study length, and ancillary cohorts such as toxicokinetic or recovery groups. Subchronic rodent studies commonly use 10 animals per sex per dose group at minimum, with additional satellites for interim sacrifice or kinetic sampling. Non-rodent studies operate with smaller cohorts but demand more intensive individual monitoring.
Underpowered designs waste animals; overpowered designs waste resources and conflict with the 3Rs principle. Calibrating numbers to the statistical question — not the convention — is the mark of a mature design process.
Minimum 10 animals per sex per dose group; satellites added for TK sampling or interim necropsies. High throughput, robust historical controls.
Smaller groups with intensive individual monitoring. Porcine, ovine, caprine, and rabbit models offer humanoid-scale organ systems for translational fidelity.
Animal numbers are justified statistically and ethically. Replacement, Reduction, and Refinement are embedded in every group-size decision from the outset.
Endpoints Evaluated in Multi-Dose Toxicology Studies
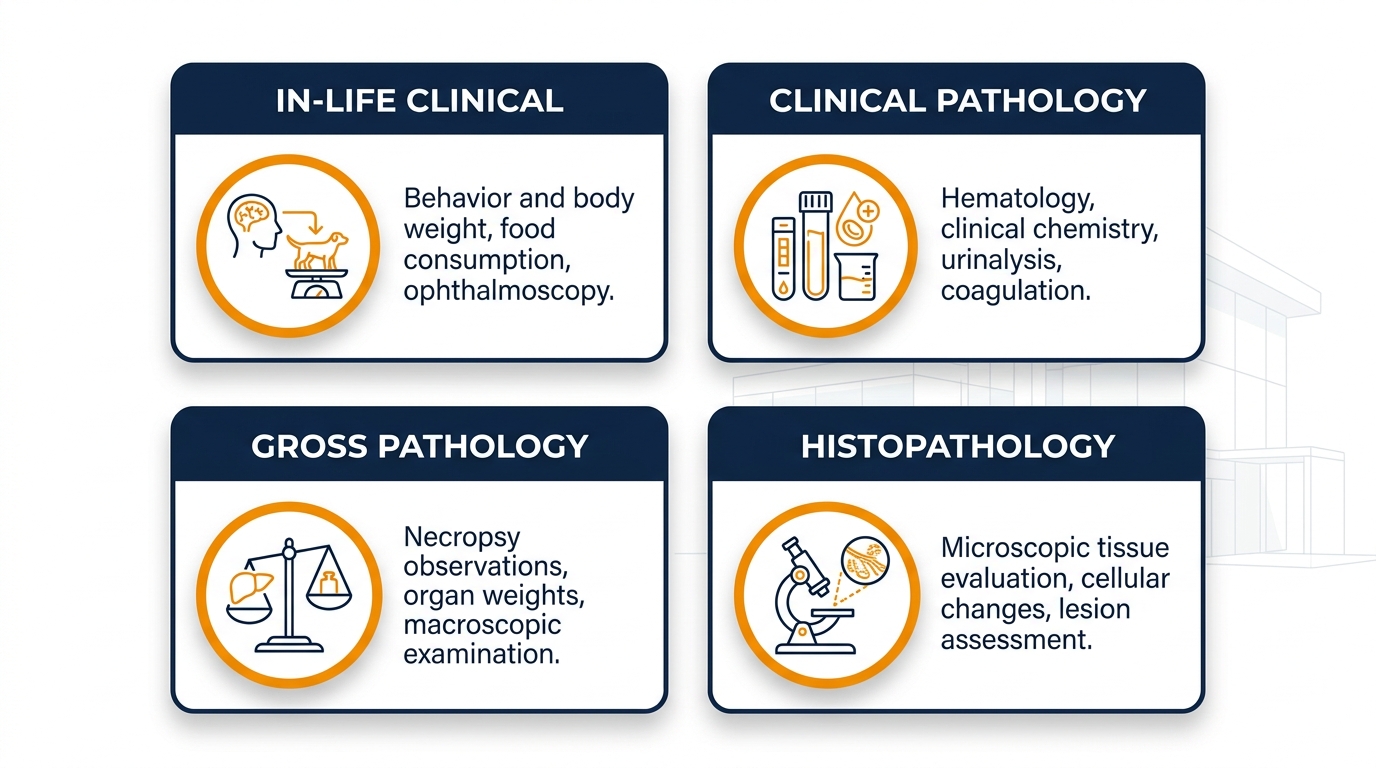
A robust repeat-dose protocol layers in-life observations onto terminal pathology to construct a coherent safety narrative. Clinical signs are scored daily, body weight and food/water consumption are tracked at defined intervals, and clinical pathology — hematology, clinical chemistry, coagulation, urinalysis — is collected at scheduled timepoints. At necropsy, gross findings, organ weights, and full histopathology of a defined tissue list are evaluated by board-certified pathologists.
| Endpoint Category | Examples | What It Reveals |
|---|---|---|
| In-life clinical | Behavior, body weight, food intake | Early systemic stress signals |
| Clinical pathology | Hematology, chemistry, urinalysis | Hepatic, renal, hematopoietic effects |
| Gross pathology | Necropsy observations, organ weights | Macroscopic target-organ involvement |
| Histopathology | Microscopic tissue evaluation | Definitive lesion characterization |
NOAEL: The Reference Point That Anchors Risk Assessment
The No Observed Adverse Effect Level is the highest tested dose at which no biologically meaningful adverse effect is detected. It functions as the practical threshold from which human exposure margins, reference doses, and starting clinical doses are derived. A defensible NOAEL depends on the quality of the underlying study — adequate dose spacing, sufficient group sizes, sensitive endpoints, and pathologist consensus on what constitutes “adverse” versus “adaptive.”
“A NOAEL produced from a poorly designed study is not a safety margin; it is a liability waiting to surface during regulatory review.”
— BIOTECH FARM Preclinical Science Team
Selecting Dose Levels: Balancing Rigor, Welfare, and Regulatory Expectation
Dose selection requires a high dose that meaningfully challenges the biological system without producing excessive morbidity, a low dose anchored near anticipated human exposure, and an intermediate dose that resolves the dose-response curve. The Maximum Tolerated Dose (MTD) often defines the upper bound, but increasing reliance on kinetically guided selection and limit-dose justifications reflects the field’s movement toward smarter, more humane designs.
The 3Rs — Replacement, Reduction, and Refinement — are not a compliance checkbox; they are a design philosophy that improves data quality.
Maximum Tolerated Dose and Its Practical Relevance
The MTD represents the ceiling at which an animal can receive the test article without unacceptable toxicity. It ensures the study probes the biological envelope sufficiently to identify hazards while staying inside ethical limits. When MTD cannot be reached due to formulation or solubility constraints, a limit dose with appropriate justification typically replaces it.
Recovery Groups: Distinguishing Reversible From Persistent Effects
A recovery cohort receives the full dosing regimen, then enters a treatment-free observation window — commonly 14 to 28 days — before terminal evaluation. This design answers two regulatory questions simultaneously: are observed toxicities reversible upon cessation, and do delayed effects emerge after exposure ends?
Recovery groups are particularly valuable for compounds with long half-lives, immunomodulatory profiles, or potential for cumulative organ stress. Including them by default in pivotal studies often saves a dedicated follow-up protocol later.
Including a recovery group by default in pivotal 90-day studies is considered best practice. The incremental cost of a satellite cohort is minimal compared to the cost of designing and executing a separate reversibility study at a later date.
Common Mistakes That Compromise Repeat-Dose Studies
Programs fail more often from execution gaps than from scientific surprises. Recurring missteps include underpowered group sizes that fail to detect moderate effects, inadequate dose spacing that flattens the response curve, poorly characterized formulations that drift in concentration over the dosing period, and clinical pathology timepoints that miss transient biomarker peaks.
- Underpowered group sizes that fail to detect moderate effects
- Inadequate dose spacing that flattens the dose-response curve
- Formulation concentration drift invalidating exposure assumptions
- Clinical pathology timepoints that miss transient biomarker peaks
- Documentation gaps — incomplete raw data or missing calibration records — that invalidate sound science during audit
Choosing a partner with rigorous SOPs and an independent quality assurance function is the single most effective hedge against these failure modes.
GLP Accreditation: Required, Not Optional, for Regulatory Submission

Good Laboratory Practice accreditation is generally required for repeat dose toxicity studies submitted to regulatory authorities in Israel and abroad. GLP enforces data integrity, traceability, and reproducibility, which is precisely what regulators rely on when accepting non-clinical safety data. In Israel, the Israel Laboratory Accreditation Authority (ISRAC) recognizes GLP-compliant facilities, and that recognition supports international acceptance of the resulting data — eliminating duplicate testing across jurisdictions.
Core Principles of GLP in Practice
GLP defines how studies are planned, performed, monitored, recorded, archived, and reported. Concretely, this means:
- Written study protocols approved prior to initiation
- Version-controlled Standard Operating Procedures
- Calibrated and qualified equipment with maintenance records
- Independent Quality Assurance Unit oversight
- Complete archived raw data with full audit trail
The OECD GLP principles via ISRAC outline the operational structure that regulatory inspectors expect to find on site.
Regulatory Compliance for Animal Studies in Israel
In Israel, repeat dose toxicity studies involving animals operate under the Animal Welfare Law (Animal Experiments), 1994, with oversight by the National Council for Experiments in Animals under the Ministry of Health. Every protocol requires ethical review and approval before initiation, and the law explicitly demands minimization of animal numbers, reduction of suffering, and replacement where validated alternatives exist.
The formal application pathway is documented through the Ministry of Health application materials, which sponsors and study directors must follow before any in-life work begins.
The Roles of ISRAC and the Ministry of Health
ISRAC recognizes GLP-compliant testing facilities, supporting international acceptance of Israeli study data. The Ministry of Health, through the National Council for Experiments in Animals, supervises ethical conduct and scientific justification of every protocol. Together, these bodies create a dual layer of oversight — technical quality and ethical legitimacy — that gives Israeli preclinical data strong standing in global submissions.
Mapping Sponsor Needs to On-Site Capabilities
Sponsors approaching a repeat-dose program rarely need a vendor; they need a scientifically supportive partner who can translate strategy into execution.
| Sponsor Need | How a Capable Facility Responds |
|---|---|
| Regulatory-grade data | Documented procedures, QA oversight, archived raw data |
| Translational relevance | Large animal models — pig, sheep, goat, rabbit — with humanoid organ scale |
| Complex surgical procedures | State-of-the-art surgery rooms, C-Arm fluoroscopy, echocardiography, 4K laparoscopy |
| Scientific dialogue | Interactive conference space, brainstorming with senior surgeons and study directors |
| Animal welfare assurance | Spacious housing, tender care practices, 3Rs-aligned design |
A repeat-dose program is a multi-month commitment in which every operational detail compounds. Partnering with a facility that combines deep preclinical experience, ethical performance, and transparent collaboration reduces protocol risk and accelerates the path to regulatory submission. BIOTECH FARM brings more than three decades of leadership in preclinical research, an advanced large animal facility, and a professional crew built for project management across cardiology, ophthalmology, orthopedics, metabolic disease, wound healing, and respiratory platforms.
“The combination of scientific escort, well-documented procedures, and animal welfare commitment supports sponsors who need data that will hold up under scrutiny — not data that will need to be repeated.”
— BIOTECH FARM Ltd.
Frequently Asked Questions
What does your regulatory pathway demand from your next preclinical safety study, and is your current design built to deliver it? Connect with the BIOTECH FARM team for a tailored discussion of protocol design, species selection, GLP execution, and on-site capabilities that match your scientific question.

This article is intended for informational purposes. For regulatory guidance specific to your program, consult qualified preclinical and regulatory affairs professionals.




