FDA Preclinical Study Guidance: A Practical Roadmap for Developers
At the heart of medical innovation, the bridge between a laboratory discovery and a first-in-human clinical trial is built on rigorous nonclinical evidence. The FDA preclinical study guidance framework defines what regulators expect to see in that evidence base — from pharmacology and toxicology to safety pharmacology, model selection, and reporting standards. Drawing on more than 30 years of experience leading and managing preclinical research on large animal models, the team at BIOTECH FARM has prepared this article to help R&D leaders translate guidance documents into a clear, risk-based plan that supports IND submissions and downstream development.
Expert Insight
The most common mistake developers make is treating preclinical planning as a checklist exercise rather than a strategic risk-reduction process. The FDA preclinical study guidance framework is flexible — and that flexibility is both an opportunity and a trap. Programs that succeed build their nonclinical package backwards from the intended first-in-human protocol, ensuring every study answers a specific regulatory or scientific question — nothing more, nothing less.
What Is FDA Preclinical Study Guidance and Who Is It For?
FDA preclinical study guidance is a collection of documents that articulates regulatory expectations for nonclinical data supporting new drugs, biologics, and medical devices. Its core purpose is twofold: ensuring sufficient evidence of safety before any human exposure, and supporting orderly progression through development phases. Key areas covered include pharmacology, pharmacokinetics, toxicology, safety pharmacology, animal model selection, and reporting standards.
The guidance is most relevant when preparing an Investigational New Drug (IND) application or equivalent regulatory submission, but it also informs internal go/no-go decisions long before the IND is filed. Sponsors of small molecules, biologics, gene/cell therapies, and combination products all rely on this framework to align scientific work with regulator expectations.
Preclinical vs. Nonclinical: Are These Terms Interchangeable?
In most regulatory contexts, “preclinical” and “nonclinical” are used almost interchangeably. However, “nonclinical” is the term preferred in formal guidance documents, and it covers any scientific work performed outside human clinical trials — including in vitro assays, ex vivo studies, and in silico modeling.
Terminology tip: Pharmacology, PK/PD, toxicology, safety pharmacology, and biocompatibility sections are labeled “nonclinical” in the eCTD structure. Internally, teams may continue to say “preclinical,” but submitted documents should follow the regulator’s vocabulary to avoid unnecessary review questions.
What Preclinical Studies Are Typically Required Before IND?
Although every program is unique, an IND-enabling package generally includes pharmacology and proof-of-concept data, a basic PK/ADME profile, and toxicology studies aligned with the proposed clinical plan. The exact scope depends on route of administration, treatment duration, the target patient population, product type, and the level of residual scientific uncertainty.
Pharmacology & POC
Mechanism of action, primary and secondary pharmacodynamics, proof-of-concept in a relevant disease model.
PK / ADME Profile
Absorption, distribution, metabolism, and excretion characterization to support dose selection and exposure-response analysis.
Toxicology Package
Repeat-dose toxicology with toxicokinetics, safety pharmacology integration, and genotoxicity battery for small molecules.
What Does an “IND-Enabling Package” Actually Mean?
An IND-enabling package is the complete set of nonclinical data — pharmacology, PK, toxicology, safety pharmacology, and CMC-linked safety information — that justifies the safety of administering an investigational drug or biologic to humans for the first time. It is the scientific foundation on which regulators decide whether the proposed clinical trial can proceed.
Does the FDA Require Animal Studies Before First-in-Human?

In vivo data are still frequently expected, but the overarching legal requirement is “sufficient evidence of safety” rather than mandatory animal studies in every situation. The FDA Modernization Act 2.0 explicitly recognizes that alternative methods can, in principle, contribute to that evidence base. The practical reality in 2024–2025 is that most IND submissions still rely heavily on animal data, supplemented — not replaced — by New Approach Methodologies (NAMs) when scientifically justified.
When Can Alternatives (NAMs) Replace Parts of the Nonclinical Package?
In vitro organ-on-chip systems, validated in silico PBPK models, and human-derived 3D tissue platforms can reduce or, in narrow cases, replace certain animal studies. Acceptance hinges on scientific validation, fit-for-purpose qualification, and a transparent risk assessment.
For general principles on the 3Rs (replacement, reduction, refinement) and considerations for animal testing applications, see the official 3Rs guidance from the Israeli Ministry of Health.
FDA Animal Study Requirements in Practice: What Is the “Minimum”?
There is no single “minimum list” published by the FDA. Instead, expectations revolve around a risk-based program. Several recurring themes are consistent across review divisions:
- Documented rationale for species and model selection
- Clear dose justification with range-finding support
- Exposure-response relationship with toxicokinetics
- Comprehensive histopathology with qualified pathologist review
- Quality documentation traceable to raw data
For more details on maintaining high-quality standards in your studies, refer to our article on Regulatory Compliance Preclinical Research GLP Standards.
How Many Species Are Needed for Toxicology — One or Two?
For small-molecule drugs, two species (typically one rodent and one non-rodent) are usually expected for repeat-dose toxicology. For biologics and human-specific products, ICH S6(R1) allows a single relevant species when scientific justification is robust.
???? Small Molecules
Two-species toxicology (rodent + non-rodent) is the standard expectation. Rat and dog, or rat and minipig, are the most common pairings.
???? Biologics / mAbs
One relevant species is often acceptable per ICH S6(R1), provided cross-reactivity and pharmacological activity are rigorously documented.
⚠️ Important: When only one species is biologically relevant, the burden of justification is high — and that justification must be defensible in writing, not just in conversation. Reviewers will scrutinize tissue cross-reactivity data, binding affinity comparisons, and pharmacological equivalency arguments.
What Is GLP and When Is It Truly Required?
Good Laboratory Practice (GLP) is a quality system governing the conduct, documentation, and quality control of nonclinical safety studies submitted to support regulatory decisions. In practice, “pivotal” toxicology and safety pharmacology studies are expected to be GLP-compliant. GLP shapes everything:
- Written SOPs covering all critical operations
- Independent Quality Assurance (QA) function
- Full traceability of raw data to final report
- Archival of records, samples, and specimens
- Complete auditability of the final study report
Early pharmacology and proof-of-concept studies are usually non-GLP, which is acceptable as long as the pivotal package is unambiguous about which studies were and were not GLP-compliant.
What Does Safety Pharmacology Include and Why Is It Critical?
Safety pharmacology evaluates undesirable effects of a candidate on vital organ systems — primarily cardiovascular, respiratory, and central nervous system function. Its purpose is to identify functional risks that toxicology alone might miss and to mitigate those risks before any human is exposed.
“Safety pharmacology data are often the deciding factor in whether a first-in-human protocol is approved as written — or sent back for additional cardiovascular monitoring. Embed these assessments early, not as an afterthought.”
— BIOTECH FARM Scientific Advisory Team
Safety pharmacology assessments can be embedded into repeat-dose toxicology studies (the “core battery integrated” approach) or run as dedicated stand-alone studies. Either approach requires the same level of methodological rigor and GLP compliance for pivotal submissions.
Acute, Repeat-Dose, and Chronic Toxicology: What Distinguishes Them?
These categories are defined by exposure duration and dosing frequency. The cardinal rule is that toxicology duration must match — or exceed — the planned human exposure duration.
| Study Type | Duration | Primary Purpose | Recovery Cohort? |
|---|---|---|---|
| Acute | Single / ≤2 weeks | MTD estimation, immediate organ effects | Rarely required |
| Repeat-Dose (Sub-acute / Sub-chronic) | 2 weeks – 3 months | NOAEL determination, target organ identification | Often included |
| Chronic | 6–12 months | Long-term organ effects, supports Phase 3 / NDA | Typically required |
The EMA’s Guideline on repeated dose toxicity (Revision 1) provides detailed regulatory expectations for this category.
Translating Animal Data to Humans: NOAEL, MTD, and HED
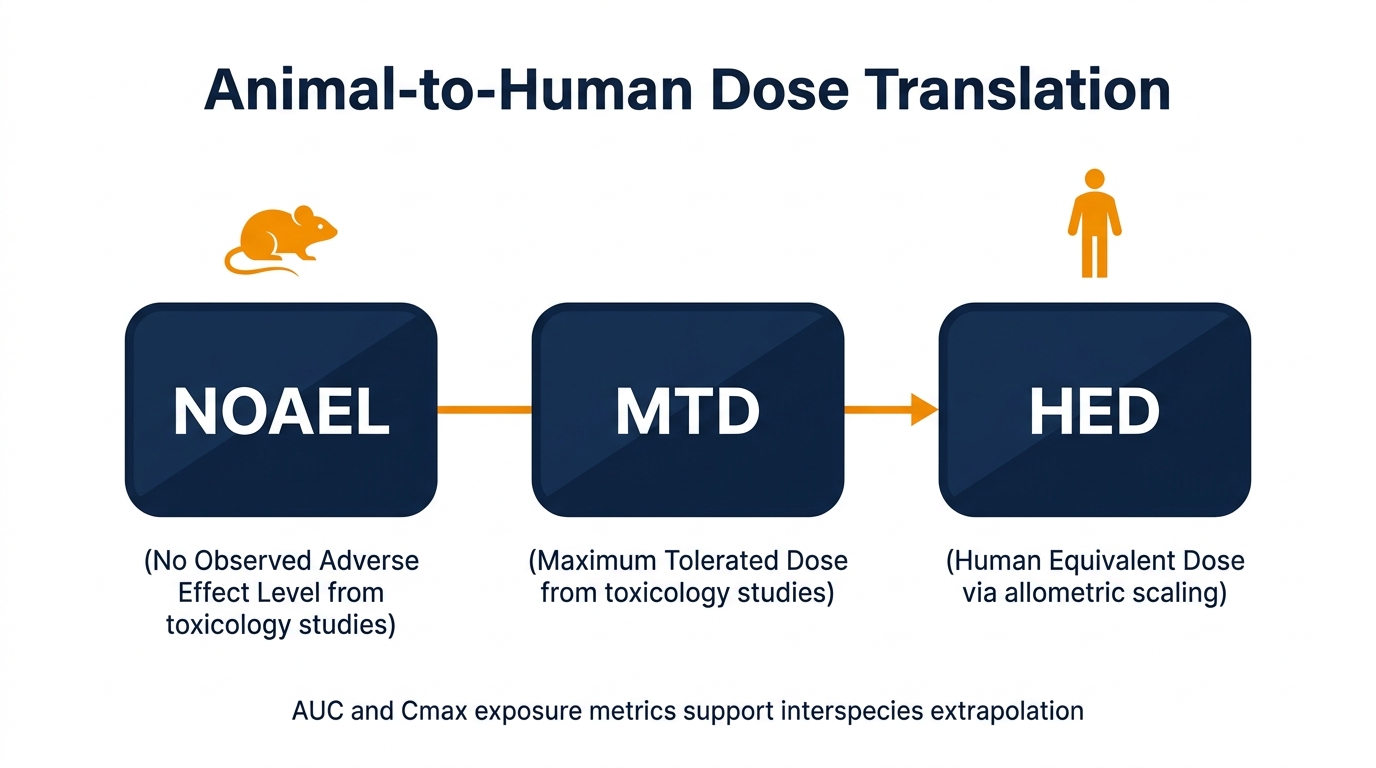
NOAEL (No Observed Adverse Effect Level), MTD (Maximum Tolerated Dose), and HED (Human Equivalent Dose) are the anchors that convert animal findings into human dosing decisions. NOAEL and MTD come from toxicology studies; HED is calculated from NOAEL using allometric scaling and a safety factor — commonly 10×, sometimes higher for biologics or sensitive populations.
Highest tested dose showing no statistically or biologically significant adverse effects — the primary anchor for human dose selection.
Maximum tolerated dose — the highest dose producing acceptable toxicity, used to define upper exposure boundaries for clinical dose escalation.
Human equivalent dose — NOAEL converted to a human starting dose using body surface area scaling and a species-specific conversion factor.
✅ Best Practice: Present systemic exposure metrics — AUC and Cmax — alongside mg/kg doses, especially when interspecies PK differences are pronounced. Exposure-based justification is consistently more persuasive to reviewers than body-weight-based extrapolation alone.
How ICH M3(R2) and ICH S6(R1) Shape FDA Preclinical Development
ICH M3(R2)
Defines the timing and scope of nonclinical studies relative to clinical trial phases and marketing approval. It functions as the planning skeleton even when the sponsor’s primary regulator is the FDA — preventing both over-testing and critical data gaps.
ICH S6(R1)
The dedicated guideline for nonclinical safety evaluation of biologics — monoclonal antibodies, recombinant proteins, fusion proteins. Emphasizes species relevance, mechanism-driven pharmacology, and accommodates one-species toxicology programs when justified.
Nonclinical Expectations by Product Type
The nonclinical requirements for small molecules, biologics, and medical devices differ substantially. Understanding which framework governs your program at the outset prevents costly misdirection:
| Aspect | Small Molecule | Biologic (mAb / Protein) | Medical Device |
|---|---|---|---|
| Primary guidance | ICH M3(R2), S7A/B | ICH S6(R1), M3(R2) | ISO 10993 series, FDA device guidance |
| Species count (tox) | Two (rodent + non-rodent) | One relevant species often acceptable | Risk-based; biocompatibility focus |
| Genotoxicity | Required (ICH S2(R1) battery) | Generally not required | Required for patient-contacting materials |
| Immunogenicity | Limited focus | Central concern | Material-dependent |
| Pivotal study GLP | Yes | Yes | Yes (per ISO 10993) |
Choosing the Right Animal Model for Preclinical Research
Model selection should be driven by biological relevance to the product’s mechanism of action and anticipated risk profile — not by what is “commonly used.” The rationale must be well-documented, scientifically defensible, and auditable. Critical considerations include:
- Target expression and cross-reactivity in the candidate species
- Disease relevance for efficacy models
- Measurable and translatable endpoints
- Reproducibility across cohorts and sites
- Anatomical approximation to human dimensions (especially for device programs)
Alternative Models: Ex Vivo, Organ-on-Chip, In Silico
Ex vivo perfused organs, organ-on-chip microphysiological systems, and in silico PBPK or QSAR models are increasingly accepted as complements to traditional animal studies. They rarely fully replace in vivo work in pivotal IND-enabling programs today, but they are powerful tools for mechanistic deconvolution, screening, and reducing animal numbers in line with 3Rs principles.
For guidance on developing preclinical plans for human-specific products like monoclonal antibodies, refer to the Generic Preclinical Development Plan from NIAID.
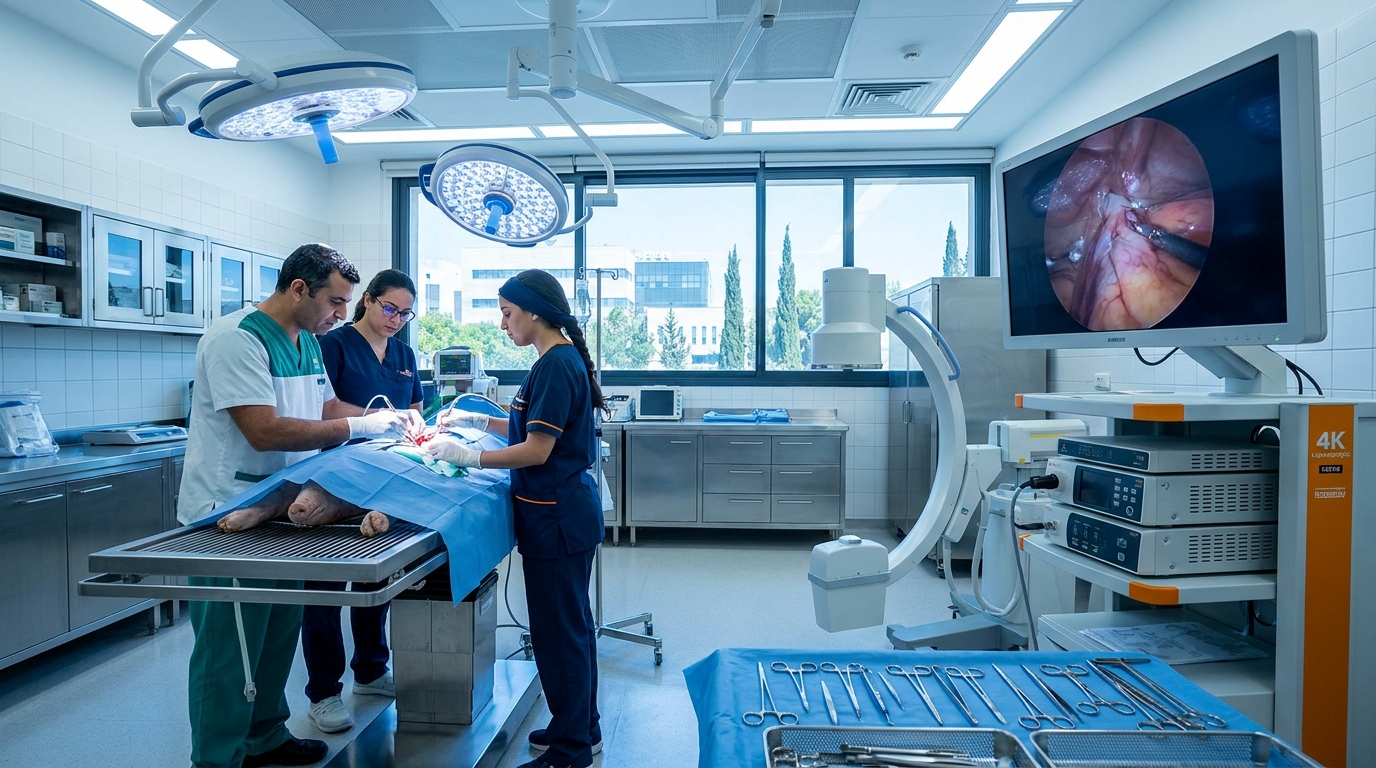
Inside the Facility: How Infrastructure Shapes Study Quality
Where a study is conducted matters as much as how it is designed. A modern large-animal facility equipped with advanced imaging and surgical technology enables faithful replication of human surgical procedures. At BIOTECH FARM, our facility includes:
Advanced Imaging
C-Arm fluoroscopy, high-definition ultrasound, cardiac echocardiography, and OCT for real-time intraprocedural guidance.
Surgical Capabilities
Surgical microscopes and 4K laparoscopic towers supporting coronary angioplasty, valve replacement, stenting, and pericardial approaches.
Animal Welfare Standards
Comfortable animal house with attentive husbandry — welfare directly influences data quality and is recognized by regulators as a quality indicator.
What Does the FDA Expect in a Nonclinical Study Report?
A nonclinical study report submitted in support of an IND or NDA must comprehensively document the study design, conduct, results, statistical analysis, deviations, and scientific interpretation. Reviewers rely on this level of detail to perform an independent risk assessment.
⚠️ Most Common Report Deficiencies
- Missing raw-data references in the final report
- Unexplained protocol deviations
- Ambiguous endpoint definitions
- Disconnect between histopathology findings and clinical interpretation
Genotoxicity and Carcinogenicity Testing: When and How
Genotoxicity (ICH S2(R1))
Standard battery for small molecules: Ames test (bacterial reverse mutation), in vitro mammalian cell assay (chromosomal aberration or mouse lymphoma TK), and in vivo micronucleus test.
Biologics acting via specific receptor interactions are typically exempt unless structural alerts justify testing.
Carcinogenicity (ICH S1)
Generally required for drugs intended for chronic human use (≥6 months continuous administration), or for products with genotoxic signals or class-related concerns.
Transgenic mouse models per ICH S1B(R1) can shorten timelines while meeting regulatory expectations.
Common Reasons the FDA Pushes Back on Nonclinical Sections
Recurring reasons for FDA information requests on nonclinical sections — and how to prevent them:
| Business Need | How a Specialized Preclinical Partner Helps |
|---|---|
| IND-enabling toxicology under tight timelines | Integrated study design, GLP-ready protocols, and scientific escort from a senior team experienced in regulator-facing programs |
| Complex large-animal surgical models | State-of-the-art surgery rooms, advanced imaging, and senior surgeons accustomed to humanoid anatomy |
| Justifying species selection for a biologic | Mechanism-led model rationale documented to ICH S6(R1) standards, with auditable records |
| Animal welfare and ethical compliance | 3Rs-aligned program (Replacement, Reduction, Refinement) with documented well-being practices |
| Cross-functional R&D collaboration | Interactive conference space and transparent project management for iterative scientific dialogue |
“Early engagement — pre-IND meetings, Type B interactions, written feedback on study designs — consistently reduces the volume and severity of late-stage pushback. The cost of a pre-IND meeting is a fraction of the cost of a major amendment six months into pivotal studies.”
— BIOTECH FARM Regulatory Strategy Team
Our Methodology: Risk-Based Nonclinical Program Design
The BIOTECH FARM approach to nonclinical program design follows a structured five-step methodology refined over two decades of IND-enabling work across cardiovascular devices, orthopedic implants, ophthalmology programs, biologics, and small-molecule pharmaceuticals:
Target Package Definition: Map the intended clinical program to define exactly what nonclinical data is needed — no more, no less.
Species and Model Selection: Select and justify the biological model using mechanism-of-action analysis and cross-reactivity data.
Protocol Design and GLP Alignment: Draft protocols with embedded welfare criteria, QA oversight plan, and auditable data management systems.
Study Execution: Conduct studies with senior veterinarians, state-of-the-art equipment, and real-time deviation management.
Report and Regulatory Support: Produce IND-ready study reports with full traceability and provide scientific support through regulatory interactions.
Frequently Asked Questions
Ready to Plan Your Nonclinical Program?
FDA preclinical study guidance, ICH M3(R2), and ICH S6(R1) together define a clear but flexible framework: build a risk-based nonclinical program, justify every choice scientifically, document everything to GLP standards where it matters, and align the package with the clinical development plan rather than with abstract checklists.
Small biotechs and established developers alike succeed when they treat the nonclinical phase as a strategic exercise — not a compliance afterthought. What is the single biggest gap between your current nonclinical plan and the IND submission you envision twelve months from now?
Regulatory Compliance Consultation
Expert guidance on FDA preclinical requirements, GLP compliance, and nonclinical strategy.
Preclinical Study Design Services
Custom study protocols built backwards from your IND submission timeline and clinical goals.
IND-Enabling Program Management
End-to-end management of your complete nonclinical package from candidate to IND filing.





