Drug Delivery Systems Preclinical Testing: Your Complete Expert Guide to Models, Decisions & Regulatory Success
With over 30 years of combined preclinical research expertise, BIOTECH FARM Ltd. has guided pharmaceutical innovators through the complex landscape of drug delivery system evaluation. Our team has witnessed firsthand how proper preclinical testing separates breakthrough therapies from costly development failures—and we’re sharing that knowledge to help you make evidence-based decisions that accelerate your path to clinical trials.
???? Expert Insight: Before a novel formulation or carrier reaches a human subject, a rigorous set of experiments must demonstrate that it meaningfully improves drug exposure, tissue targeting, efficacy, or safety compared to conventional approaches. Without robust preclinical evidence, development resources are wasted on candidates that fail predictably in later stages.
What Decisions Does Preclinical Testing for Drug Delivery Systems Actually Support?
Drug delivery systems preclinical testing is not a single experiment—it is an integrated program designed to answer specific, critical questions that determine whether your investment yields returns or becomes a cautionary tale.
Critical Questions Your Preclinical Program Must Answer:
- Does the carrier improve bioavailability compared to free drug?
- Does it deliver more drug to the target tissue while reducing systemic exposure?
- Is the release profile consistent with the intended dosing regimen?
- Are there unexpected safety signals from the carrier itself?
The answers to these questions drive pivotal decisions: selecting a lead formulation from several candidates, defining dose and frequency for pivotal studies, justifying animal model choices to regulators, and ultimately building the data package that supports an Investigational New Drug (IND) application.
Which Preclinical Endpoints Matter Most When Evaluating a Drug Delivery Platform?
Evaluating a drug delivery platform requires measuring more than just plasma drug concentrations. The endpoints that matter most span pharmacokinetics, biodistribution, pharmacodynamics, and safety. Each provides a different lens through which a platform’s value can be assessed.
| Endpoint Category | Key Measurements | Why It Matters |
|---|---|---|
| Systemic PK | AUC, Cmax, Tmax, half-life, clearance | Quantifies overall drug exposure and absorption kinetics |
| Biodistribution | Tissue-to-plasma ratio, organ accumulation | Confirms whether the carrier delivers drug to the target site |
| PK/PD Relationship | Target engagement markers, biomarker kinetics | Links exposure to biological response |
| Safety / Local Tolerance | Injection site reactions, histopathology, hematology | Identifies carrier-related toxicity early |
| Carrier Characterization | Release/degradation rate, particle integrity | Verifies in vivo behavior matches in vitro design |
⚠️ Critical Warning: Without measuring biodistribution alongside PK, a sponsor may observe improved AUC but have no evidence that more drug actually reached the intended tissue. Similarly, omitting pharmacodynamic endpoints makes it difficult to demonstrate that a delivery platform offers a clinical advantage beyond simply altering the PK profile.
How Do You Choose the Right Animal Model for Testing Drug Delivery?
Selecting the wrong animal model is one of the fastest routes to generating non-translatable data. Model choice depends on several interrelated factors: the intended administration route, the target organ’s anatomical and physiological similarity across species, the drug’s clearance mechanism, and whether a disease model is required.
Screening programs frequently begin with rodent models for initial PK and tolerability assessments, then progress to larger species—such as pigs, sheep, or rabbits—when the anatomy of the target organ, the volume of sampling required, or the administration device demands it. Large animal models provide data that are often more directly translatable to human physiology, particularly for cardiovascular, orthopedic, and ophthalmic drug delivery platforms.
Criteria Checklist for Model Selection
Before committing to an in vivo study, teams should systematically evaluate:
- Route feasibility: Can the formulation be administered as intended in the chosen species?
- Sampling volume: Will sufficient blood or tissue be available without compromising the animal?
- Target tissue accessibility: For sampling or imaging
- Immunogenicity risk: From the carrier or drug
- Disease relevance: When efficacy endpoints are included
BIOTECH FARM’s experience with animal models in preclinical research demonstrates how matching these criteria to available species prevents costly study redesigns.
Healthy vs. Disease Models—When Each Is Justified
Healthy animal models are typically appropriate for PK, biodistribution, and safety studies where the primary question is “where does the drug go and at what concentration?” Disease models become necessary when the study objective requires demonstrating efficacy or when pathology alters drug distribution—for instance, tumor models that exhibit the enhanced permeability and retention (EPR) effect relevant to nanoparticle targeting.
Using a disease model for a PK-only question adds unnecessary complexity and ethical burden; conversely, using a healthy model to demonstrate efficacy of a targeted delivery system may miss the translational mark entirely.
— BIOTECH FARM Research Team
A Typical Preclinical Study Flow for Novel Drug Delivery Systems
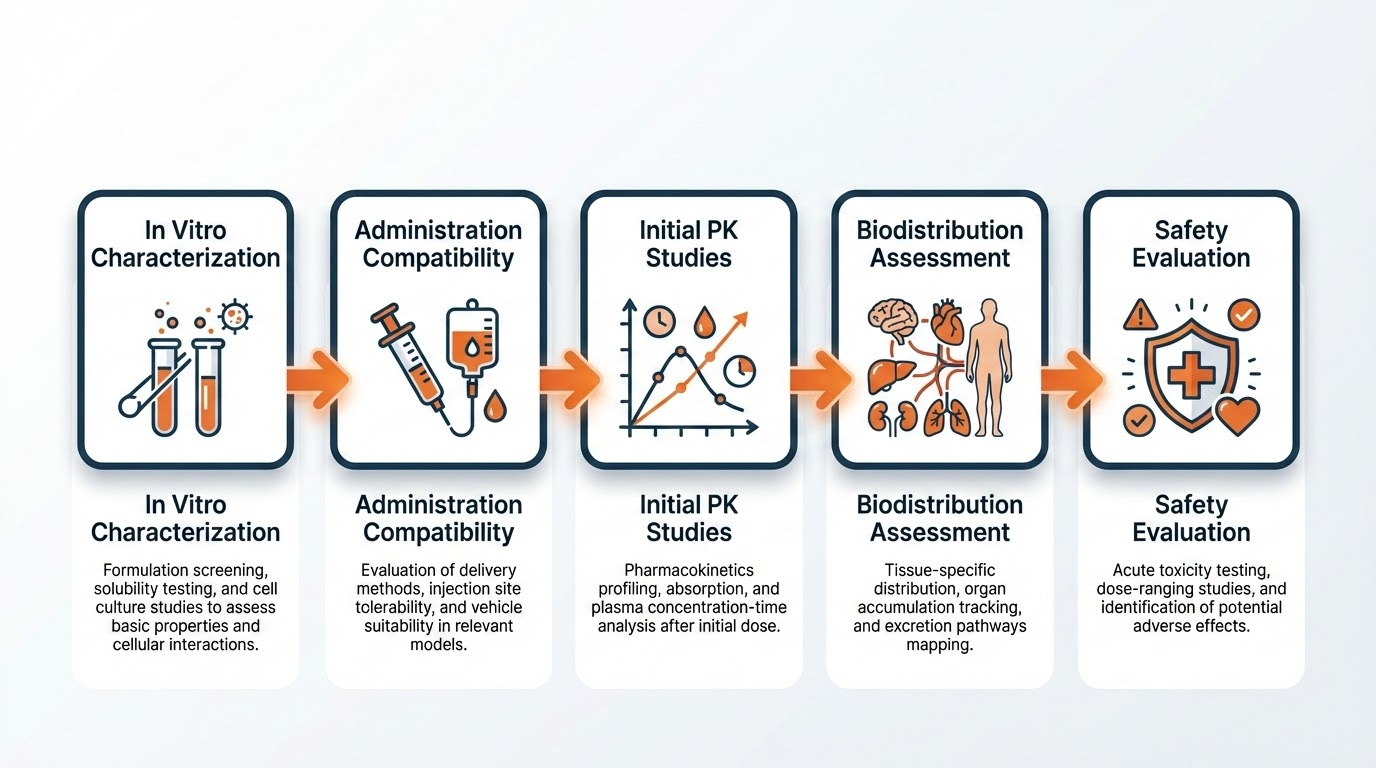
While no two development programs are identical, a general study flow has emerged as best practice:
Standard Study Sequence:
- Step 1: In vitro characterization (stability, release kinetics, particle properties)
- Step 2: Administration compatibility testing
- Step 3: Initial PK studies establishing concentration-time profile
- Step 4: Biodistribution assessments
- Step 5: PK/PD or efficacy studies (if targeting specific disease indication)
- Step 6: Safety and local tolerance evaluations
- Step 7: GLP pivotal studies (or reformulation if needed)
Important: The order adapts to risk. A well-characterized carrier with a history of safe use may allow safety studies to occur later, whereas a first-in-class nanoparticle platform may demand local tolerance data before any multi-dose PK work.
What In Vitro Tests Are Expected Before In Vivo Dosing?
⚠️ Common Mistake: Rushing into animal studies without adequate in vitro data is a common and preventable mistake that wastes resources and delays programs.
Before any in vivo dosing, the formulation should have:
Formulation Characterization
- ✓ Documented stability data
- ✓ Release/dissolution profile
- ✓ Particle size distribution (if applicable)
- ✓ Zeta potential & polydispersity index
Route-Specific Parameters
- ✓ Osmolality & viscosity
- ✓ pH confirmation
- ✓ Sterility (for injectables)
- ✓ Analytical method compatibility
Prevents These Failures
- ✗ Precipitation in syringe
- ✗ Needle blockage
- ✗ Dose variability from sedimentation
- ✗ Quantification problems
Testing Controlled Release or Sustained Release In Vivo—What Could Go Wrong?
Demonstrating sustained release in an animal model requires more than showing a flat concentration-time curve. The study must differentiate genuine controlled release from the carrier from slow absorption due to depot effects at the injection site—a phenomenon known as “flip-flop” kinetics.
Sampling Schedule Design to Capture the Tail
For sustained-release formulations, the sampling window must extend well beyond the expected duration of release. Missing the terminal phase of the PK profile—the “tail”—leads to underestimation of AUC and incorrect half-life calculations.
Common Pitfalls in Sustained-Release Studies
- Adsorption artifacts: Drug or carrier adsorbing to collection materials (tubes, syringes, filters)
- Sample degradation: Compromised data integrity during storage or processing
- Insufficient group sizes: Inter-animal variability obscuring the release profile (groups of 3 often inadequate)
- Missing reference data: No IV control for absolute bioavailability comparison
How Is Biodistribution Measured in Preclinical Drug Delivery Studies?
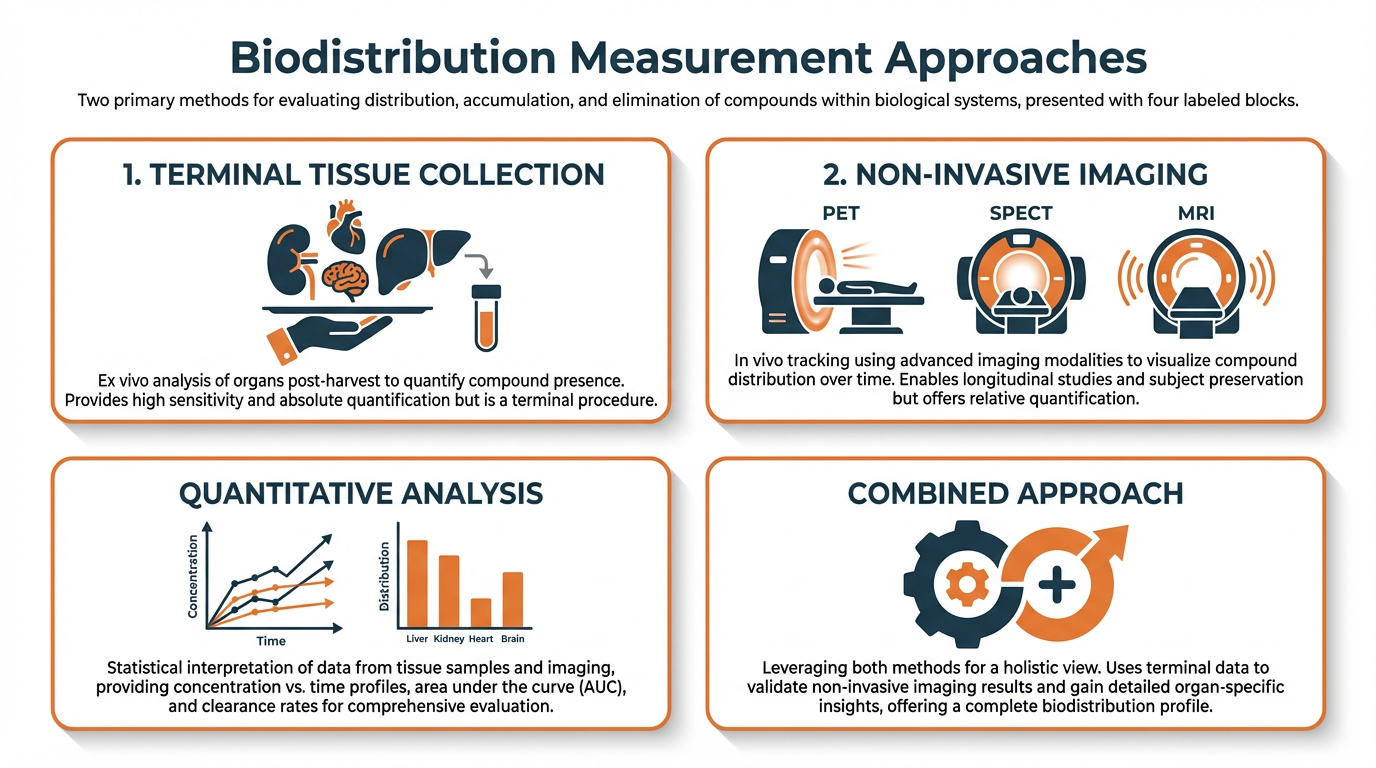
Biodistribution data answer the fundamental question of targeted delivery: did the carrier actually change where the drug goes?
| Approach | Advantages | Limitations |
|---|---|---|
| Terminal Tissue Collection | Direct, quantitative data; gold standard | More animals needed; only snapshots at each time point |
| Non-Invasive Imaging (PET, SPECT, MRI, optical) | Dynamic data; fewer animals; spatial & temporal kinetics | Tracks label not drug; potential label dissociation |
✓ Best Practice: Combining both approaches strengthens the evidence package considerably. Facilities equipped with advanced imaging alongside established tissue processing capabilities ensure that biodistribution claims rest on robust, multi-method evidence.
Distinguishing PK, TK, and PD in Drug Delivery Evaluation
These three abbreviations appear constantly in preclinical discussions, but their distinctions matter profoundly for drug delivery systems:
Pharmacokinetics (PK)
Drug concentrations over time—absorption, distribution, metabolism, and elimination
Toxicokinetics (TK)
Same measurements within toxicity study context—establishing dose-exposure-toxicity relationships
Pharmacodynamics (PD)
Biological or therapeutic response to the drug—the “what happens” after exposure
For drug delivery platforms, linking PK to PD is especially important. A carrier that changes the PK profile is only valuable if that change translates into a measurable improvement in efficacy, duration of action, or reduction in toxicity. Without PD or biomarker endpoints, the preclinical dataset demonstrates a “different” profile but cannot demonstrate a “better” one.
When Is PK/PD Modeling Worth the Investment?
PK/PD modeling becomes particularly valuable when sponsors need to:
- Select among multiple dosing regimens
- Compare formulations quantitatively
- Demonstrate an exposure-response relationship that justifies clinical progression
- Simulate scenarios requiring prohibitively many animal studies
Evaluating Dosing Routes: Why IV Reference Data Are Non-Negotiable
Route selection for a drug delivery system aligns with the clinical target—subcutaneous for patient self-administration, intranasal for CNS access, intramuscular for depot formulations—but IV administration is nearly always tested as a reference.
IV data provide absolute bioavailability and define the drug’s intrinsic PK without absorption-related variables, creating the baseline against which all other routes are compared.
| Route | Formulation Constraints | Key Safety Endpoints |
|---|---|---|
| Subcutaneous | Specific pH, tonicity, viscosity ranges | Injection site reactions, local tolerance |
| Intranasal | Address mucociliary clearance | Mucosal irritation, ciliary function |
| Intramuscular | Appropriate viscosity, particle size for injection | Local tolerance, depot behavior |
| Intravenous | Sterility, osmolality, particle-free | Infusion reactions, hemolysis |
BIOTECH FARM’s capabilities in drug delivery models and PK determination across multiple administration routes in large animals provide sponsors with the flexibility to evaluate these parameters in a single facility.
Why Drug Delivery Systems Fail in Preclinical Translation
Understanding common failure modes helps teams design studies that address weaknesses early:
Stability Issues
Degradation of carrier or premature drug release during storage or administration
Poor Scale-Up
Variability from laboratory to manufacturing batches that preclinical data cannot predict
Unexpected Toxicity
From carrier components, off-target accumulation, or immunogenicity against carrier material
Model Mismatch
Chosen rodent model poorly represented human physiology for target organ or route
Efficacy-Toxicity Trade-off
Excellent target-site concentrations but unacceptable systemic side effects
Missing Go/No-Go Data
Major resources committed before generating decisive data points
Each of these failure points can be mitigated—though not eliminated—by thorough in vitro characterization, thoughtful model selection, and phased study designs that generate go/no-go data before major resource commitments.
Regulatory Expectations for Preclinical Drug Delivery Studies

Regulatory agencies including the FDA and EMA expect that preclinical programs for drug delivery systems provide clear justification for the formulation chosen, the animal models used, and the study designs employed. The ICH M3(R2) guideline serves as the foundational framework for nonclinical safety studies required before and during human clinical trials.
When Is GLP Required?
Good Laboratory Practice (GLP) compliance is generally required for pivotal toxicology studies whose results will be submitted to regulatory authorities as part of an IND or marketing authorization application.
Exploratory, dose-finding, and proof-of-concept studies are often conducted under non-GLP conditions, provided they are well-documented and scientifically sound.
Key Resources:
Working with a preclinical facility that understands both GLP and non-GLP study requirements—and maintains documentation standards that facilitate a smooth transition between them—reduces regulatory risk significantly.
Comparing Two Drug Delivery Systems Head-to-Head: A Practical Framework
When sponsors have multiple candidate formulations, preclinical comparison studies must be designed with scientific rigor to ensure meaningful results.
Head-to-Head Comparison Essentials:
- PK parameters: AUC, Cmax, Tmax under matched conditions
- Biodistribution profiles: Target tissue vs. off-target accumulation
- Efficacy: In a relevant model if available
- Safety/local tolerance: Carrier-specific effects
- Unified analytical methods: Essential across all test articles
- Reference control: Typically IV solution of free drug
- Randomization & blinding: Where feasible
The deliverable from such a study should include not just raw numbers but a clear interpretive framework that enables a rational decision between candidates.
What Deliverables Should a Preclinical CRO Provide?
A high-quality preclinical contract research organization should deliver a comprehensive package:
| Deliverable | Description |
|---|---|
| Study Plan (Protocol) | Detailed experimental design with endpoints and analysis plan |
| Raw Data | Auditable format with full traceability |
| Detailed Study Report | Methods, results, statistical analysis, scientific interpretation |
| Analytical Data | Representative chromatograms or spectra |
| QA Statements (GLP) | Quality assurance documentation for regulated studies |
| Regulatory Summaries | Tailored for IND or other submissions |
Data integrity and traceability are non-negotiable. Every data point should be traceable from the final report back to the original instrument output. BIOTECH FARM’s commitment to transparency in collaboration and well-documented procedures ensures that sponsors receive deliverables suitable for regulatory scrutiny.
How Drug Delivery Systems Facilitate Targeted Delivery
Targeted delivery is the aspirational endpoint for many novel drug delivery platforms. The concept involves engineering carriers—nanoparticles, liposomes, antibody-drug conjugates, hydrogels—to accumulate preferentially at a specific anatomical site or bind to specific cellular receptors.
Passive Targeting
Exploits physiological phenomena such as the EPR effect, where nanoparticles accumulate in tumors due to leaky vasculature
Active Targeting
Incorporates ligands—peptides, antibodies, aptamers—on the carrier surface to engage specific receptors overexpressed on target cells
Preclinically, demonstrating targeted delivery requires quantitative biodistribution data showing differential accumulation, not merely in vitro binding assays. For a deeper exploration, see this review of nanocarriers for the delivery of medical, veterinary, and agricultural active ingredients.
New Approach Methodologies: Can They Replace Animal Studies?
New Approach Methodologies (NAMs)—encompassing advanced in vitro systems, organ-on-chip platforms, computational models, and in silico simulations—represent a growing component of the preclinical toolkit.
Current Status: Regulatory acceptance of NAMs as full replacements for animal studies remains endpoint- and drug-type-specific.
The FDA’s framework for streamlined nonclinical studies and acceptable NAMs outlines where these methodologies may be integrated. Currently, NAMs are most valuable as complementary tools that inform study design, reduce the number of in vivo experiments needed, and provide mechanistic context for animal data.
Advanced Bioanalytical Techniques for Drug Delivery System Characterization
Accurately quantifying a drug, its metabolites, and the carrier material in complex biological matrices requires sophisticated bioanalytical methods:
| Technique | Application | Key Advantage |
|---|---|---|
| LC-MS/MS | Small molecule quantification | Gold standard sensitivity and selectivity |
| ICP-MS | Inorganic nanoparticle-based systems | Metal content quantification in tissues |
| Autoradiography | Spatial drug distribution | Tissue-level localization |
| Mass Spectrometry Imaging | Drug distribution within tissues | Spatial information without radiolabeling |
Method validation for sensitivity, selectivity, accuracy, precision, and matrix effects is essential; unvalidated methods produce data that neither the sponsor nor the regulator can trust.
How BIOTECH FARM Supports Your Drug Delivery Program
| Practical Need | How BIOTECH FARM Supports It |
|---|---|
| Multiple administration routes in a single study | Large animal facility equipped for IV, SC, IM, intranasal, and specialized routes with experienced surgical and veterinary teams |
| Comprehensive PK sampling over extended windows | Large animals allow serial blood sampling with sufficient volume, reducing animal numbers while capturing full concentration-time profiles |
| Advanced imaging for biodistribution | State-of-the-art imaging tools including fluoroscopy and high-definition ultrasound, integrated with tissue collection capabilities |
| Regulatory-ready documentation | Transparent collaboration with well-documented procedures, supporting both non-GLP and GLP study frameworks |
| Scientific guidance throughout the program | Over 30 years of experience in preclinical R&D with senior scientists providing scientific escort from study design through reporting |
For further insights into ethical frameworks and practical strategies in animal studies, see Preclinical Animal Studies: Key to Medical Breakthroughs.
Frequently Asked Questions
Ready to Design Your Drug Delivery Preclinical Program?
Have you identified the critical endpoints, the right animal model, and the regulatory framework for your novel drug delivery system—but need a partner with the facilities, expertise, and scientific depth to execute?
BIOTECH FARM’s team brings over three decades of preclinical R&D experience, advanced large animal surgical and imaging capabilities, and a tailored approach that matches your specific program needs.




