Dose Range Finding Study: A Practical Guide to DRF and MTD in Preclinical Research
Translating a promising molecule from the bench into a viable clinical candidate depends on one critical bridge: well-designed preclinical dose-finding work. A dose range finding study (DRF) is the early decision engine that defines what doses make sense, where toxicity begins, and how exposure relates to effect. With more than three decades of combined experience leading and managing preclinical research on large animal models, the team behind BIOTECH FARM approaches DRF and MTD studies as scientifically supportive partners — combining rigorous protocol design, transparent documentation, and humane standards to give sponsors a defensible foundation for subsequent GLP studies, IND/CTA submissions, and first-in-human trials.
🔎 Expert Insight
A well-conducted DRF study is not simply a regulatory checkbox — it is the most cost-effective investment a sponsor can make before committing to a GLP pivotal study. By clearly defining the dose-response landscape, integrating TK/PK data, and pre-specifying tolerability criteria, teams can avoid the costly scenario of an uninterpretable or repeated GLP study. At BIOTECH FARM, every DRF is designed as a decision-making tool that directly and defensibly informs what comes next.
What Is a Dose Range Finding Study and Why Is It Conducted in Preclinical Research?
A dose range finding study is an early-stage preclinical toxicology or pharmacology investigation whose primary goal is to identify suitable dose levels for subsequent, more definitive studies — typically pivotal GLP toxicity studies — and to inform initial human dosing. By mapping a probable dose range, candidate target organs, and the relationship between systemic exposure (TK/PK) and emerging toxicology, a DRF dramatically reduces uncertainty before sponsors commit resources to expensive pivotal work.
DRF studies are typically short, employ several dose groups, and guide downstream decisions on dose selection, dosing schedules, sampling strategy, and the choice of endpoints. A well-conducted DRF saves money, accelerates timelines, and protects animal welfare by preventing poorly designed pivotal studies that may need to be repeated.
How Does a DRF Study Differ From an MTD Study?
A DRF study maps dose ranges and dose-response trends across multiple dose groups, while an MTD study is specifically designed to identify the maximum tolerated dose based on predefined toxicity and tolerability criteria. In practice, the two often overlap: many programs combine elements, designing a DRF that also yields an MTD estimate.
Some sponsors use a DRF primarily to select three dose levels (low/mid/high) for a repeat-dose study, ensuring the high dose produces “sufficient but non-lethal” toxicity. Others run a dedicated MTD study where escalation continues until clear tolerability limits emerge. The choice depends on program objectives, regulatory expectations, and the molecule’s known pharmacology.
What Is “Tolerability” Versus “Toxicity” in the Context of MTD?
Tolerability describes the degree to which an organism withstands the test article without unacceptable adverse effects, while toxicity refers to the adverse effects themselves — which become intolerable above certain dose thresholds. Common factors used to define tolerability in MTD work include body weight loss (often a 10–20% threshold), severity of clinical signs, clinical pathology shifts in hematology and chemistry, and predefined humane endpoints. Clear, written tolerability criteria established before the study begins are essential for consistent decision-making.
DRF Study Focus
- Maps full dose-response range
- Identifies candidate target organs
- Informs dose selection for GLP
- Multiple dose groups with comparisons
MTD Study Focus
- Defines tolerability ceiling
- Escalation until defined limit reached
- Predefined stopping/reduction rules
- Can be combined with DRF design
How Is Dose Escalation Determined in Animal Studies?
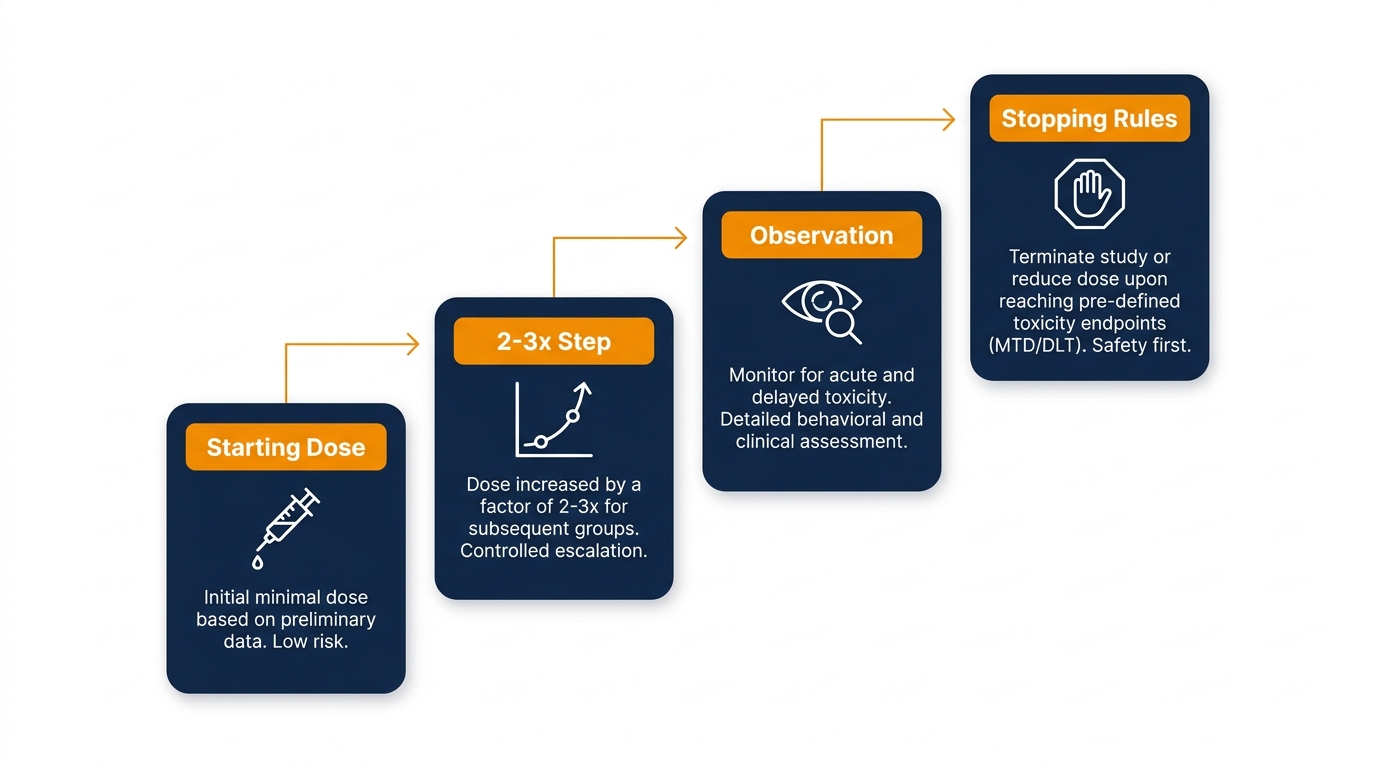
Designing dose escalation in animal studies typically uses a staggered scheme with logarithmic steps — often 2- to 3-fold increases — that cover a wide range efficiently while allowing observation between cohorts. The goal of a dose escalation animal study is to reach a dose that produces expected toxicity without crossing ethical limits or generating data that cannot be interpreted.
Predetermined stopping or reduction rules are essential and usually include thresholds for significant body weight loss, severe clinical signs, marked laboratory changes (e.g., transaminases, hematologic shifts), and any mortality or humane euthanasia. Staggered escalation also helps identify a clear “breaking point” and avoids irrelevant dose jumps that waste animals and resources. According to the OECD humane endpoints guidance, clinical signs should be systematically recognized and used to trigger humane intervention.
📌 Protocol Design Note
Predetermined stopping rules must be written into the protocol before study initiation — not decided post hoc. This single practice protects both scientific integrity and regulatory defensibility, and is a hallmark of the discipline BIOTECH FARM applies to every DRF design.
How Is the Maximum Tolerated Dose (MTD) Defined in Preclinical Research?
The maximum tolerated dose (MTD) is the highest dose of a test article that can be administered without producing severe or unacceptable toxicity or undue mortality, judged against predefined protocol criteria. Critically, the MTD is always study-dependent — it is shaped by treatment duration, dosing frequency, route of administration, species and strain, and the humane standards of the testing facility.
In preclinical practice, MTD is typically determined by integrating clinical signs, body weight loss, clinical pathology trends, and (when collected) macroscopic and histopathological findings. Because the definition is contextual, two studies on the same molecule may yield different MTD values if their designs differ — which is why protocol-level criteria must be transparent.
“The MTD is not a fixed biological property of a molecule — it is a contextual endpoint defined by study design, species, route, and the rigour of predefined tolerability criteria.”
— BIOTECH FARM Preclinical Research Team
MTD Versus NOAEL: Why the Distinction Matters for IND/CTA
MTD represents a tolerability ceiling, while NOAEL (No Observed Adverse Effect Level) is the highest dose at which no adverse effects are observed under the study conditions. NOAEL is the conventional starting point for deriving the initial human dose, combined with exposure considerations (HED conversion) and a safety factor — typically 10 — as described in the FDA guidance on Maximum Safe Starting Dose.
A well-designed DRF ensures that the subsequent GLP study covers a range allowing both clear NOAEL identification and observable toxicity at the high dose. A high dose set too low risks missing relevant toxicity for IND or CTA review; a high dose set too high may invalidate the study with excessive toxicity that cannot be interpreted.
⚠️ Critical Distinction for Regulatory Submissions
MTD and NOAEL serve different regulatory functions. Confusing the two — or failing to clearly document which dose level fulfilled which criterion — is a recurring cause of IND/CTA queries. Every DRF report from BIOTECH FARM explicitly maps dose levels against both concepts, providing reviewers with a clear, transparent decision trail.
What Endpoints Are Evaluated in a DRF Study Preclinical?
Common endpoints in a DRF study preclinical include clinical observations, body weight and food consumption, clinical pathology (hematology and clinical chemistry), and — when scientifically justified — necropsy and organ weights. Optional inclusion of TK/PK sampling links systemic exposure to observed toxicity, often the most informative addition for downstream decisions.
Endpoint selection should reflect anticipated risk and the molecule’s mechanism of action: candidates with cardiac liabilities warrant ECG and cardiac biomarker monitoring; CNS-active molecules call for neurobehavioral observations; ocular products demand specialized ophthalmic exams. The ICH S3A guidance outlines how TK should be integrated into toxicology studies, including pilot and DRF data.
Is Histopathology Always Required in DRF?
Histopathology is not always mandatory in a DRF. For many programs, a lighter set of in-life and clinical pathology tools is sufficient to support dose selection. However, for products with known target organ concerns, novel modalities, or unusual administration routes (e.g., intraocular, intrathecal, implanted devices), histopathology can be decisive and should be designed in from the start.
📋 Core Endpoints
- Clinical observations & signs
- Body weight & food consumption
- Hematology & clinical chemistry
- Necropsy & organ weights (when justified)
📈 Optional/Specialized Endpoints
- TK/PK sampling & bioanalysis
- ECG & cardiac biomarkers
- Ophthalmic / neurobehavioral exams
- Histopathology (target organ concern)
Balancing Animal Numbers in DRF With the 3Rs Principle
The number of animals in a DRF depends on the study’s specific goal — selecting GLP doses, identifying MTD, integrating TK — and the expected biological variability of the model. DRF designs are deliberately leaner than GLP studies, using the minimum number of groups and animals required to make a defensible decision.
Strategies to minimize animal use under the 3Rs framework (Replacement, Reduction, Refinement) include microsampling for TK, staggered cohort designs, sentinel groups, and early stopping rules. The EMA Q&A on ICH S3A highlights how analytical advances and microsampling reduce or eliminate satellite TK groups.
✅ 3Rs Best Practice at BIOTECH FARM
BIOTECH FARM integrates 3Rs thinking at every stage of DRF design: microsampling strategies are discussed during protocol development, sentinel group designs are considered for high-risk first-in-species studies, and humane endpoint criteria are written into every protocol before study initiation — not added retrospectively.
How Long Does a Dose Range Finding Study Take?
A typical dose range finding study timeline spans preparation (protocol design, logistics, animal procurement, formulation qualification), a short in-life phase ranging from days to a few weeks depending on dosing duration, followed by analytical work and reporting. End-to-end duration is usually a few weeks to several months.
Key time-contributing factors include availability of animal models, analytical method development and qualification for TK, formulation complexity, and the number of sampling time points. Programs that integrate logistics with the testing facility from the start — including animal acclimation, dosing rehearsal, and bioanalytical readiness — typically compress timelines without compromising data quality.
🕐 Typical DRF Study Phase Breakdown
- Protocol & Preparation: Animal procurement, formulation qualification, method validation — typically 2–6 weeks
- In-Life Phase: Dosing, clinical observations, sampling — days to several weeks
- Analysis & Pathology: Bioanalysis, clinical pathology, histopathology — 2–4 weeks
- Reporting & Dose Decision: Final report, dose selection for GLP — 2–4 weeks
Is a DRF Study Required to Be GLP?

In most programs, a DRF study is a non-GLP supporting investigation. Pivotal toxicology studies submitted for regulatory submission must be GLP compliant, but DRFs are decision-making tools rather than pivotal records. That said, designing a non-GLP DRF with high quality and documentation standards — traceable raw data, defined acceptance criteria, recorded deviations, and transparent dose-decision logic — strengthens the program substantially and protects sponsors during later regulatory review.
For context on facility recognition principles, see the official Israeli accreditation page on OECD-GLP recognition of testing facilities. A facility that operates with GLP-aligned discipline even on non-GLP work delivers a significantly more defensible package.
Selecting Low, Mid, and High Doses for a Repeat-Dose Study After DRF
After a DRF, dose selection for the repeat-dose study typically follows a logical pattern: the low dose targets NOAEL or a relevant pharmacological exposure, the mid dose provides an intermediate margin to characterize the dose-response, and the high dose approaches MTD or tolerable toxicity. Influencing factors include observed clinical signs, clinical pathology trends, systemic exposure metrics (AUC/Cmax), and technical feasibility — solubility, dose volume, and route tolerability. The OECD TG 407 standard provides a formal framework for 28-day repeated-dose oral toxicity.
| Dose Group | Selection Goal | Key Inputs from DRF |
|---|---|---|
| Low | Establish NOAEL / minimal pharmacological exposure | Clean clinical observations, baseline TK |
| Mid | Characterize dose-response slope and margin | Mild reversible findings, intermediate AUC |
| High | Approach MTD / observable but non-lethal toxicity | Defined toxicity signals, exposure plateau check |
What If MTD Is Not Reached?
If MTD is not reached due to solubility, exposure plateau, or formulation constraints, alternative strategies include defining a limit dose, modifying the route or formulation, or shifting to a maximum feasible dose criterion. This approach is recognized in regulatory guidance such as ICH S1C(R2) and is particularly relevant for biologics with saturable targets.
Why Integrating TK/PK Into DRF Improves Dose Decisions
Integrating TK/PK into a DRF establishes the link between actual exposure and observed toxicity, identifies non-linearity or saturation, and allows dose selection based on exposure rather than mg/kg alone — significantly reducing the risk of an uninterpretable GLP study.
If toxicity emerges without a corresponding rise in exposure, this may indicate a different mechanism or absorption limitation; if exposure jumps disproportionately between mid and high doses, dose intervals likely need adjustment. The OECD TG 417 Toxicokinetics guideline formalizes how TK data support these dose decisions.
“A DRF study without TK is like navigating with a map but no scale — you can see the terrain, but you cannot judge the distances that matter most for your GLP design.”
— BIOTECH FARM Scientific Advisory Perspective
Common Mistakes in DRF/MTD Planning and How to Avoid Them
Several recurring planning mistakes weaken DRF and MTD studies and undermine the resulting GLP work. A too-narrow dose range misses the boundaries needed for proper dose selection; a too-wide range wastes animals on irrelevant cohorts. Undefined or vague MTD criteria force subjective judgment after the fact. Insufficient TK sampling leaves exposure-response questions unanswered.
Other frequent errors include “chasing MTD at all costs” when exposure has plateaued — generating mortality without informative data — and selecting a high dose that causes severe suffering, rendering the study ethically and scientifically unusable.
| Mistake | Consequence | Mitigation |
|---|---|---|
| Narrow dose range | Missed MTD or NOAEL | Use logarithmic spacing across ≥3 levels |
| Vague MTD criteria | Subjective stopping decisions | Predefine weight, clinical, and lab thresholds |
| Sparse TK sampling | Cannot link exposure to toxicity | Plan AUC/Cmax coverage; use microsampling |
| Formulation overlooked | Infeasible high dose | Qualify formulation before in-life start |
DRF for Biologics Versus Small Molecules: What Changes?
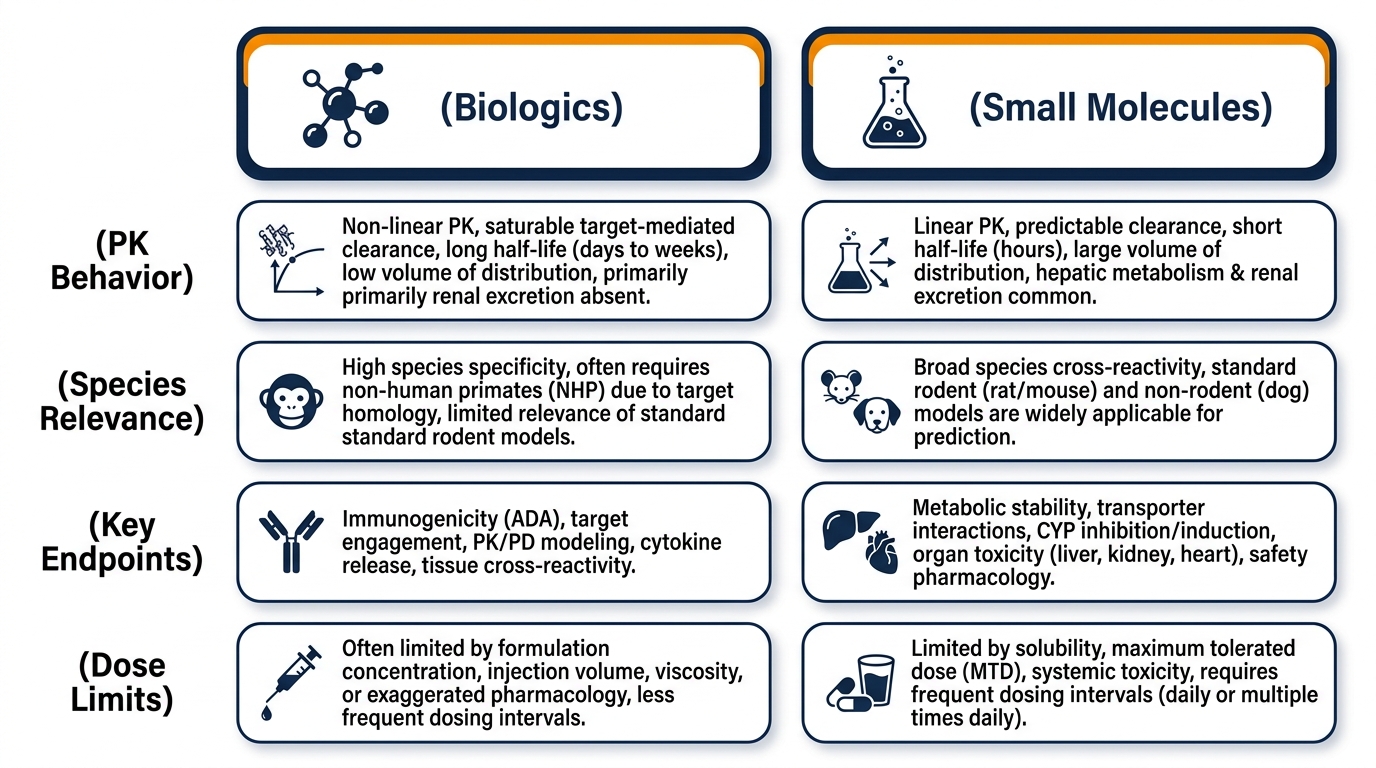
A dose range finding study for biologics and one for small molecules share the same strategic purpose but differ substantially in design. Biologics often display saturable targets and non-linear PK, carry immunogenicity risks (anti-drug antibodies, ADAs) that affect PK/PD and toxicity, and may be limited by production capacity or dose volume. Species selection becomes pivotal — non-rodent or large-animal models are frequently required for target relevance.
Small molecules generally show more linear PK, with metabolism playing a larger role in dose-response; safety margins are often wider and species/route choice more flexible, with traditional toxicology endpoints (organ weights, histopathology, clinical pathology) as standard. Both pathways share core priorities: careful dose selection, characterization of dose-response and exposure-response relationships, and full commitment to animal welfare.
| Aspect | Biologics | Small Molecules |
|---|---|---|
| PK behavior | Often non-linear, saturable | Typically more linear |
| Species relevance | Critical; often non-rodent / large animal | Greater flexibility |
| Key endpoints | ADAs, PD biomarkers, target engagement | Organ weights, clinical chemistry, histopathology |
| Dose limits | Volume / production constraints | Solubility / formulation constraints |
How BIOTECH FARM Supports DRF and MTD Studies in Practice
Translating these principles into a real study requires more than a protocol — it requires an integrated facility, an experienced surgical and veterinary team, and transparent project management. The BIOTECH FARM facility is built around large animal models (pig, sheep, goat, rabbit) with state-of-the-art surgery rooms equipped with C-Arm fluoroscopy, high-definition ultrasound, echocardiography, and 4K laparoscopic towers — enabling sophisticated DRF designs that include cardiovascular, ophthalmic, orthopedic, and respiratory endpoints.
The animal house is designed for high-quality husbandry and ethical performance, with humane endpoints, refined handling, and documented procedures supporting the 3Rs. Scientific escort from senior investigators ensures that protocol design, dose-decision logic, and TK integration are aligned with the sponsor’s regulatory pathway from the earliest planning stage.
Mapping Sponsor Needs to Facility Capabilities
| Sponsor Need | How the Facility Supports It |
|---|---|
| Defensible dose selection for GLP | Tailored DRF protocols with predefined criteria and full traceability |
| Complex large-animal models | Pig, sheep, goat, rabbit with humanoid organ relevance |
| Integrated TK/PK and imaging endpoints | On-site surgery suites with fluoroscopy, ultrasound, OCT, microscopy |
| Ethical, transparent execution | Documented humane endpoints, refined husbandry, ethics-led oversight |
| Responsive, collaborative project management | Scientific escort, interactive planning, brainstorming environment |
Frequently Asked Questions





