Preclinical to Clinical Translation: Bridging Bench, Bedside, and Beyond
Translating preclinical findings into safe and effective human therapies is one of the most demanding scientific journeys in modern medicine. At BIOTECH FARM, decades of hands-on experience with large animal models, GLP-aligned procedures, and integrated scientific escort allow research teams to design preclinical programs that anticipate clinical realities rather than react to them. The result is a more disciplined pathway from molecule or device prototype toward first-in-human (FIH) studies, supported by transparent documentation and rigorous animal welfare standards.
Expert Insight
True translational preclinical research is not a single experiment with a positive readout in an animal model — it requires alignment of mechanism of action, therapeutic exposure, validated biomarkers, and clinical endpoints across species. Every dataset should have a pre-defined role in informing the next decision gate, from IND-enabling toxicology through first-in-human dose selection.
What is Preclinical to Clinical Translation?
Preclinical to clinical translation is the structured process of converting in vitro and in vivo findings into informed clinical decisions: which patients to enroll, how to dose, what to measure, and which safety signals to monitor. True translational preclinical research is not a single experiment with a positive readout in an animal model; it requires alignment of mechanism of action, therapeutic exposure, biomarkers, and clinical endpoints across species.
The NCATS overview frames this as the T0–T4 spectrum, where bench to bedside preclinical work sits at the heart of risk reduction. BIOTECH FARM contributes domain expertise in translational medicine animal models for medical devices and pharmaceuticals, providing infrastructure that supports decision-quality data.
Why Does the Preclinical Clinical Gap Persist?

The preclinical clinical gap arises because animal systems, however valuable, simplify human biology. Standard cohorts are typically young, healthy, single-sex, genetically uniform, and housed in controlled microenvironments — conditions that rarely match real patient populations with comorbidities, polypharmacy, and age-related variability.
Immune responses, metabolism, and disease heterogeneity all introduce uncertainty when readouts are extrapolated to humans. Recognizing the gap as a structural property of the system — rather than a failure of any single study — encourages investigators to plan for it: by diversifying models, validating biomarkers across species, and pre-defining how preclinical data will inform clinical decisions.
Key contributing factors to the gap: genetic uniformity of laboratory animals, controlled housing conditions, absence of comorbidities and polypharmacy, and the challenge of scaling pharmacological exposure across species with different metabolic profiles.
What is the “Valley of Death” in Drug Translation?
The valley of death describes the stage where scientifically promising candidates stall before or during the T1 transition into human subjects. Common reasons include the absence of predictive biomarkers, inability to define a safe and pharmacologically meaningful starting dose, gaps in toxicology coverage, and operational or regulatory feasibility issues.
Programs may also fail because mechanism evidence in animals does not translate into measurable clinical signal at achievable exposures. Addressing this region of risk demands deliberate translational preclinical research: front-loaded mechanistic studies, exposure-response characterization, and reverse-translation from the intended clinical context back into animal study design before commitments to expensive late preclinical work.
- Absence of predictive, validated biomarkers
- Inability to define a safe starting dose
- Incomplete toxicology coverage
- Mechanism not translating to measurable clinical signal
Why Do Drugs Succeed Preclinically but Fail in Clinical Trials?
Failures in clinical trials often originate upstream. Non-predictive animal models, flawed study design, inappropriate scaling of dose, or endpoints lacking clinical relevance are common contributors. Selection bias, lack of blinding or randomization, underpowered cohorts, and opportunistic time-point selection inflate apparent effect sizes and erode reproducibility, as highlighted in the influential Nature commentary on preclinical research standards.
Translational medicine animal models that look impressive in isolation may collapse under the heterogeneity of human trial populations. Robust preclinical to clinical translation therefore requires methodological rigor equal to that expected in late-phase clinical research.
“Improving the quality of preclinical research requires a culture shift — from treating animals as mere test vessels toward disciplined experimental science with the same standards applied to human trials.”
— Nature, Preclinical Research Standards Commentary
How Can You Tell if an Animal Model is Human-Relevant?
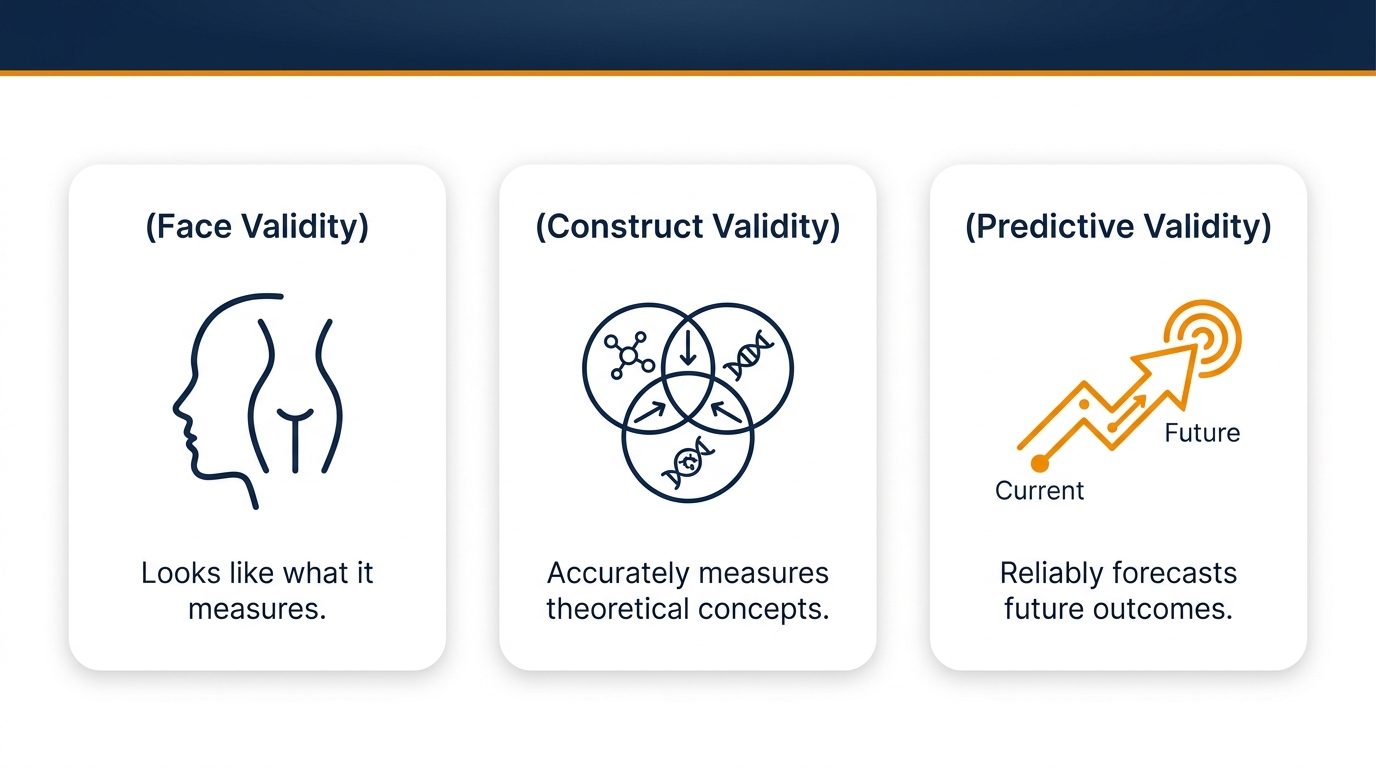
A human-relevant model reproduces disease mechanism, biological pathways, and measurable endpoints that map onto human physiology. Three forms of validity guide assessment: face validity (phenotypic resemblance), construct validity (shared underlying pathology), and predictive validity (capacity to forecast efficacy or toxicity). Validating biomarkers across species — confirming that a target engagement marker behaves comparably in rodent, large animal, and human samples — strengthens translational confidence.
Without this layered validation, even well-conducted experiments risk producing data that cannot be acted upon clinically.
Practical Criteria for Selecting an Animal Model
Selection should weigh genetic similarity, anatomical and physiological resemblance, disease progression timing, and the model’s ability to reproduce hallmark features of the human condition. Large animal models — including pig, sheep, and rabbit — often provide humanoid organ scale and procedural fidelity that small rodents cannot match, particularly for cardiology, ophthalmology, and orthopedic device evaluation.
Phenotypic resemblance to human disease presentation — does the model look like the clinical condition?
Shared underlying pathology — does the model share the biological mechanism driving the human disease?
Capacity to forecast efficacy or toxicity in humans — does a positive result in the model predict clinical success?
Efficacy, Effectiveness, and Translatability: A Comparison
These three concepts are often conflated, yet they describe distinct properties that influence translational decision-making. A model with strong efficacy but weak translatability — for example, requiring exposures unattainable in humans — provides limited value for downstream decisions.
| Concept | Setting | Question Answered | Translational Implication |
|---|---|---|---|
| Efficacy | Idealized lab/animal conditions | Does it work under controlled conditions? | Necessary but not sufficient evidence |
| Effectiveness | Real-world or complex models | Does it work under realistic variability? | Improves likelihood of clinical relevance |
| Translatability | Cross-species/clinical | Will preclinical results predict human outcomes? | Determines program viability |
What Does Translational Preclinical Research Look Like in Practice?
Translational preclinical research is proactive, designed from the outset around the clinical question. Investigators define the target patient population, plausible dosing windows, the response measurement strategy, and anticipated safety risks before the first animal is enrolled. The objective is to assemble an evidence chain linking proposed mechanism, target engagement, exposure, and clinical-relevant endpoints.
This bench to bedside preclinical orientation reduces wasted effort and ensures every dataset has a defined role in informing IND-enabling work or device feasibility submissions.
- Clinical question defined before first animal enrolled
- Target patient population and dosing windows specified
- Evidence chain linking mechanism → exposure → endpoint
- Every dataset has a defined role in go/no-go decisions
Designing Bench to Bedside Preclinical Studies Correctly
Effective design begins with the clinical endpoint and works backward. Identifying the desired Clinical Target Product Profile (TPP) clarifies which preclinical experiments will most reduce uncertainty. Structured decision gates and clear go/no-go criteria support disciplined resource allocation, as discussed in the NCATS 2015 report on the translational science spectrum.
Reverse-Translation Targeting the Clinical Target Product Profile
Defining the TPP early — efficacy thresholds, route of administration, dosing frequency, target population — gives preclinical scientists the criteria against which study designs are calibrated. Models, endpoints, and exposure ranges are then selected to test whether the candidate can plausibly meet the TPP, not merely whether it produces any biological effect.
Decision Gates and Go/No-Go Criteria Roadmap
Pre-specified milestones convert ambiguous datasets into actionable decisions. Each gate evaluates whether the accumulated evidence justifies progression, redesign, or termination. This discipline reduces sunk-cost bias and protects programs from advancing on weak foundations.
- Define Clinical TPP before model selection
- Map clinical endpoints to preclinical equivalents
- Pre-specify go/no-go criteria at each gate
- Establish exposure targets tied to human PK projections
- Justify model selection against all three validity types
Which Preclinical Study Design Errors Most Harm Translation?
Recurring methodological flaws — absent randomization, lack of blinding, underpowered samples, opportunistic endpoint selection, and incomplete reporting — inflate effect sizes and undermine reproducibility. P-hacking and selective time-point analysis can transform noise into apparent signal. The ARRIVE Guidelines 2.0 provide concrete recommendations that materially improve preclinical to clinical translation.
Quality Checklist: Randomization, Blinding, Power, Pre-Registration
A practical checklist includes: randomized allocation with documented method, blinded outcome assessment, a priori power calculation tied to a clinically meaningful effect size, pre-registered protocol with primary and secondary endpoints, and pre-specified inclusion/exclusion criteria. Together these elements protect against the most common sources of inflated findings.
How to Reduce Bias Without Overburdening Operations
Standard operating procedures, automated data capture where feasible, independent analysts, and electronic randomization codes reduce bias without inflating cost. Embedding these practices into the facility’s default workflow — rather than treating them as study-specific add-ons — keeps overhead manageable while strengthening data quality.
How to Improve Reproducibility for Better Clinical Translation
Reproducibility improves through standardized procedures, transparent reporting, appropriate controls, and replication across sites where feasible. Detailed SOPs, calibrated equipment, and full disclosure of protocol deviations matter as much as the headline result. Reporting frameworks curated by the EQUATOR Network support consistent disclosure across journals and regulatory submissions.
BIOTECH FARM emphasizes well-documented procedures and transparency in collaboration as part of standard project management, allowing sponsors to audit, reproduce, or extend studies with minimal friction.
- Detailed SOPs embedded in default facility workflow
- Calibrated equipment and full deviation disclosure
- Transparent audit-ready study documentation
- ARRIVE 2.0 and EQUATOR-aligned reporting standards
Selecting Preclinical Endpoints That Translate to Clinical Endpoints
Endpoints should reflect functional outcomes or core pathophysiology rather than convenient surrogate markers without demonstrated clinical linkage. Imaging, functional assays, and translational biomarkers with direct human equivalents strengthen the predictive value of preclinical readouts and reduce the leap of faith required at the clinical interface.
Endpoint Mapping: Biological → Functional → Clinical
For example, an enzymatic activity reduction (biological) may produce measurable organ function improvement (functional), which in turn correlates with symptom reduction or improved quality-of-life scores in patients (clinical). Constructing this chain explicitly during planning ensures each preclinical measurement contributes to a clinically interpretable narrative.
(Biological)
(Functional)
(Clinical)
What Are Translational Biomarkers, and Why Are They Critical?
Translational biomarkers are measurable indicators that can be assessed in both preclinical models and human subjects. They serve as bridges for mechanism of action, target engagement, exposure-response relationships, and disease progression. PD biomarkers confirm pharmacological activity, surrogate endpoints provide early efficacy signals, and safety biomarkers flag emerging toxicity.
Without robust translational biomarkers, dose selection and patient stratification in early clinical trials rest on weaker foundations.
Confirm pharmacological activity and target engagement at the relevant exposure level.
Provide early efficacy signals before definitive clinical outcomes are observable.
Flag emerging toxicity early, enabling adaptive monitoring in FIH study designs.
How Is Dose Translated from Animals to Humans?

Dose translation is rarely a simple weight-based extrapolation. Sophisticated pharmacokinetic and pharmacodynamic modeling, allometric scaling, and exposure-based comparisons (AUC, Cmax) inform a defensible starting dose. Regulatory frameworks rely on the No Observed Adverse Effect Level (NOAEL) from animal studies, converted to a Human Equivalent Dose (HED) with safety factors applied, as described in FDA and EMA guidance on starting-dose estimation and first-in-human risk mitigation.
When Weight-Based Scaling is Misleading
Simple mg/kg conversions ignore interspecies differences in metabolism, plasma protein binding, receptor density, and clearance pathways. Two species achieving the same mg/kg dose may experience radically different exposures and pharmacological effects.
How PK/PD Reduces Dose Uncertainty
PK/PD modeling integrates concentration-time data with biological response, enabling exposure-targeted dose selection rather than dose-targeted exposure guessing. This significantly reduces the risk of selecting a starting dose that is either subtherapeutic or unsafe.
What is PK/PD, and Why is it Key for Translation?
PK (pharmacokinetics) describes what the body does to the drug — absorption, distribution, metabolism, excretion. PD (pharmacodynamics) describes what the drug does to the body — pharmacological and toxicological effects. Integrating PK/PD establishes a quantitative link between exposure and response that is foundational to translational medicine animal models.
If the preclinical pharmacological effect requires exposures unattainable in humans, the program faces structural risk regardless of efficacy data, and replanning is preferable to advancement.
“If the preclinical pharmacological effect requires exposures unattainable in humans, no amount of impressive animal data changes the clinical prognosis. PK/PD integration is the earliest and most cost-effective risk filter available to translational scientists.”
— Adir Koreh, CEO, BIOTECH FARM Ltd.
QSP and Quantitative Models: Closing the Gap
Quantitative Systems Pharmacology (QSP) and related modeling frameworks integrate biological knowledge, PK/PD data, and experimental observations to simulate drug response across doses, schedules, and patient subgroups. They support hypothesis testing without exhausting in vivo resources and help identify scenarios where translation is most likely to succeed. For broader context on the role of in vivo evidence in this ecosystem, see the overview of Animal Models in Preclinical Research.
Essential Data for Useful Models, Not “Decoration”
Predictive QSP requires high-quality inputs: in vitro potency and binding parameters, preclinical PK/PD profiles, disease pathophysiology data, and human physiological constants. Models built on sparse or low-quality data produce visually compelling but operationally weak predictions.
Combining In Vitro, Ex Vivo, and In Vivo into One Translational Plan
A coherent program treats experimental scales as a continuum of evidence rather than independent silos. Each level should be designed to inform the next, building a cumulative argument that justifies clinical entry.
| Scale | Examples | Translational Role |
|---|---|---|
| In vitro | Cell cultures, biochemical assays | Lead identification, mechanism elucidation |
| Ex vivo | Organoids, patient biopsies | Bridging cellular and tissue-level biology |
| In vivo | Rodent and large animal models | Systemic effects, safety, integrated efficacy |
Large animal facilities equipped with imaging tools such as fluoroscopy, echocardiography, and laparoscopic platforms enable in vivo studies that approach the procedural realism of clinical practice.
What Are GLP Studies, and When Are They Required?
Good Laboratory Practice (GLP) is a quality system governing non-clinical safety studies — toxicology, safety pharmacology, and supporting pharmacokinetics — intended for regulatory submission. GLP-aligned conduct is required when data will support an IND, CTA, or equivalent application to authorities such as the FDA, EMA, or the Israeli Ministry of Health.
International standards, including ICH M3(R2) and OECD test guidelines, define the technical and procedural expectations. Programs that anticipate GLP requirements early avoid costly rework when transitioning from exploratory to pivotal studies.
GLP governs non-clinical laboratory studies supporting regulatory submissions. GCP governs the conduct of clinical trials in humans. Both are quality systems, but they apply to different stages of the development continuum with distinct documentation and oversight requirements. Anticipating GLP needs at the exploratory phase — not only the pivotal phase — is a hallmark of experienced translational programs.
How Many Animal Studies Are Needed Before First-in-Human Trials?
The required scope depends on the product type (small molecule, biologic, device), intended indication, duration of human exposure, and regulatory pathway. Typical packages include pharmacology and PK/PD studies, safety pharmacology covering vital systems, and toxicology studies (acute, repeated-dose, genotoxicity, and reproductive or carcinogenicity studies when relevant). ICH M3(R2) provides the international reference for timing and scope.
Before first-in-human dosing, sponsors typically complete general toxicology in two relevant species (one rodent, one non-rodent) to identify target organs and establish NOAELs; safety pharmacology covering cardiovascular, respiratory, and central nervous systems; genotoxicity assessments; and pharmacokinetic characterization. Large animal models with humanoid organ scale are particularly valuable for device-oriented safety assessment, where procedural realism shapes both safety conclusions and operator training.
- General toxicology — two species (rodent + non-rodent)
- Safety pharmacology — cardiovascular, respiratory, CNS
- Genotoxicity assessment
- Pharmacokinetic characterization across test species
- NOAEL determination → Maximum Recommended Starting Dose
Regulatory Landscape for Preclinical to Clinical Translation in Israel
In Israel, the Ministry of Health regulates clinical trials in alignment with ICH-GCP standards. Helsinki Committees review and oversee trials to safeguard ethical conduct and patient safety. For sponsors planning preclinical work in Israel, BIOTECH FARM offers familiarity with local regulatory expectations alongside international standards, easing the path from animal studies to multicenter clinical engagement.
- Ministry of Health oversight aligned with ICH-GCP standards
- Helsinki Committee ethical review for all human trials
- Health regulations on human experimentation and Ministry procedures
- BIOTECH FARM familiar with both local and international regulatory expectations
How BIOTECH FARM Supports Translational Programs
The following table maps common sponsor needs to operational capabilities that support preclinical to clinical translation at BIOTECH FARM.
| Sponsor Need | How the Facility Supports It |
|---|---|
| Procedural realism for device studies | Large animal models with humanoid organ scale; equipped surgery rooms with C-Arm, ultrasound, and 4K laparoscopic towers |
| Decision-quality data for go/no-go gates | Scientific escort, well-documented procedures, transparent reporting |
| Regulatory readiness | Familiarity with GLP-aligned conduct and Israeli/international regulatory expectations |
| Ethical and welfare standards | Adherence to the 3Rs, animal welfare protocols, and ethical performance |
| Tailored study design | Matching needs and services to the program’s clinical target product profile |
Frequently Asked Questions
Ready to Plan Your Next Translational Program?
How will your next preclinical study be designed to answer a specific clinical question, rather than only an experimental one? If you are evaluating models, endpoints, or regulatory strategy for a drug or medical device program, the BIOTECH FARM team is available to discuss tailored study design, large animal capabilities, and scientific escort throughout the project.





