Implant Safety Study Animals: The Backbone of Preclinical Device Validation
Understanding the rigorous process of implant safety study animals is paramount for medical device developers aiming to bring safe and effective implantable devices to market. These in vivo investigations form the scientific bridge between bench-level material characterization and first-in-human use, providing data that no laboratory dish or computer simulation can fully replicate.
20+ Years Large Animal Expertise
ISO 10993 & GLP-Aligned Protocols
End-to-End Scientific Escort
For implantable devices — whether orthopedic screws, vascular stents, neural electrodes, or absorbable scaffolds — the host’s biological response unfolds across days, weeks, and months. Animal studies allow developers to capture acute surgical response, chronic tissue remodeling, and long-term mechanical behavior in a physiologically relevant environment. These studies are also a regulatory expectation under FDA biological evaluation pathways, EU MDR clinical evaluation routes, and the ISO 10993 series. Every protocol is ethically grounded: animal use must be justified, suffering minimized, and the absence of validated alternatives demonstrated.
What Is an Implant Safety Study in Animals?
An implant safety study in animals is a preclinical investigation in which a medical device, material, or prototype is surgically placed into a living animal model to evaluate biological response, performance, and durability over a defined period. It sits at the core of implantable device testing and implant preclinical evaluation, generating evidence that informs benefit–risk decisions before human exposure.
The core objective is twofold: characterize the local tissue reaction at and around the implantation site, and screen for systemic biological responses that could indicate broader safety concerns. Findings feed directly into regulatory submissions — IDE, 510(k), De Novo, PMA, or MDR technical documentation — and shape labeling, intended use, and risk management files.
Our expertise in preclinical studies ensures comprehensive medical device safety evaluations that meet global regulatory requirements while integrating GLP-aligned documentation, surgical realism on large animal models, and end-to-end scientific escort from protocol design through final report.
Why Are Animal Models Essential for Implantable Device Testing?
Only in vivo models can simulate the complex biological environment and long-term interactions of an implant within a living system. Implantable device testing demands that researchers observe how blood, immune cells, fibroblasts, and surrounding tissues actually behave around a foreign body — not just whether a material is cytotoxic in a flask.
In vitro assays cannot fully capture chronic inflammation, fibrous encapsulation dynamics, wound healing cascades, mechanical loading at the tissue–device interface, or systemic effects from leachables distributed via circulation. The local response evolves through distinct phases — acute inflammation, chronic inflammation, foreign body reaction, and fibrous encapsulation — a sequence well documented in the scientific literature on host response to implants.
Physiological Fidelity
Living systems provide immune cascades, vascular responses, and mechanical loading that no in vitro model can replicate for permanent implants.
Regulatory Requirement
FDA, EU MDR, and ISO 10993 frameworks expect in vivo data for permanent or long-term implants where no alternative generates equivalent evidence.
Temporal Completeness
Multiple timepoints capture the full biological arc from acute surgery through chronic steady-state — essential for credible long-term safety claims.
Biocompatibility vs. Implant Preclinical Evaluation: What’s the Difference?
These terms are often used interchangeably, but they describe different scopes of work. Biocompatibility testing focuses on the biological risk posed by the material itself — its chemistry, leachable components, degradation products, and direct effects on cells and tissues. Implant preclinical evaluation is broader: it covers biocompatibility but also functional performance, deliverability, surgical handling, mechanical durability, and integration in a physiological environment.
In practical terms, biocompatibility is a subset of the overall preclinical evaluation. A device can be made of well-characterized, biocompatible materials yet still fail clinically due to migration, mechanical fatigue, or unintended tissue interaction. The FDA’s basics of biocompatibility framework underscores that material safety is necessary but not sufficient — functional and procedural endpoints matter as well.
“A device can be made of entirely biocompatible materials and still fail clinically — implant preclinical evaluation captures performance, integration, and durability that material testing alone cannot.”
— BIOTECH FARM Ltd. Scientific Team
Understanding the Chronic Implant Study
A chronic implant study evaluates the long-term safety and performance of a device by leaving it implanted for an extended duration — weeks, months, or in some cases years. It is a critical component of long-term implant safety assessment, especially for permanent or long-dwell devices such as orthopedic implants, cardiovascular prostheses, and neuromodulation systems.
The study’s purpose is to identify delayed or cumulative adverse reactions: late-onset inflammation, capsule maturation, material degradation, particulate release, mechanical wear, corrosion, or gradual functional drift. For devices intended for permanent contact, regulators typically expect data spanning multiple timepoints to confirm that the host–device interface stabilizes and that no unexpected late events emerge after the acute healing phase has resolved.
One of the most frequent missteps in implant preclinical evaluation is selecting duration based on what “looks standard” rather than on a documented risk rationale. Reviewers increasingly expect justification linking chosen timepoints to the device’s intended use, contact duration, expected degradation profile, and known failure modes. A 90-day study for a permanent implant, with no rationale for omitting longer endpoints, is a predictable deficiency.
How Long Does a Long-Term Implant Safety Study Typically Last?
Study duration is driven by the device’s intended clinical exposure and risk profile. Common ranges include 28 days for short-term contact assessments, 12–13 weeks for medium-term, and 26, 39, or 52 weeks for long-term implants — with some studies extending beyond a year for permanent devices or biodegradable materials with slow resorption profiles.
Timepoints — both early and late — are chosen to differentiate acute surgical response from chronic remodeling and steady-state biology. The FDA’s guidance on the use of ISO 10993-1 ties endpoint selection and study duration directly to cumulative time of contact, reinforcing that duration is a risk-based decision rather than a fixed default.
How are study timepoints chosen to optimize efficiency and cost?
Timepoint selection considers device novelty (newer materials warrant more frequent sampling), anticipated failure modes (early timepoints capture deployment issues; later ones capture wear, corrosion, or fatigue), and ethical use of animals. Imaging and non-terminal sampling allow longitudinal data from the same animal, reducing total numbers while preserving statistical power.
Choosing the Right Animals for Your Implant Safety Study
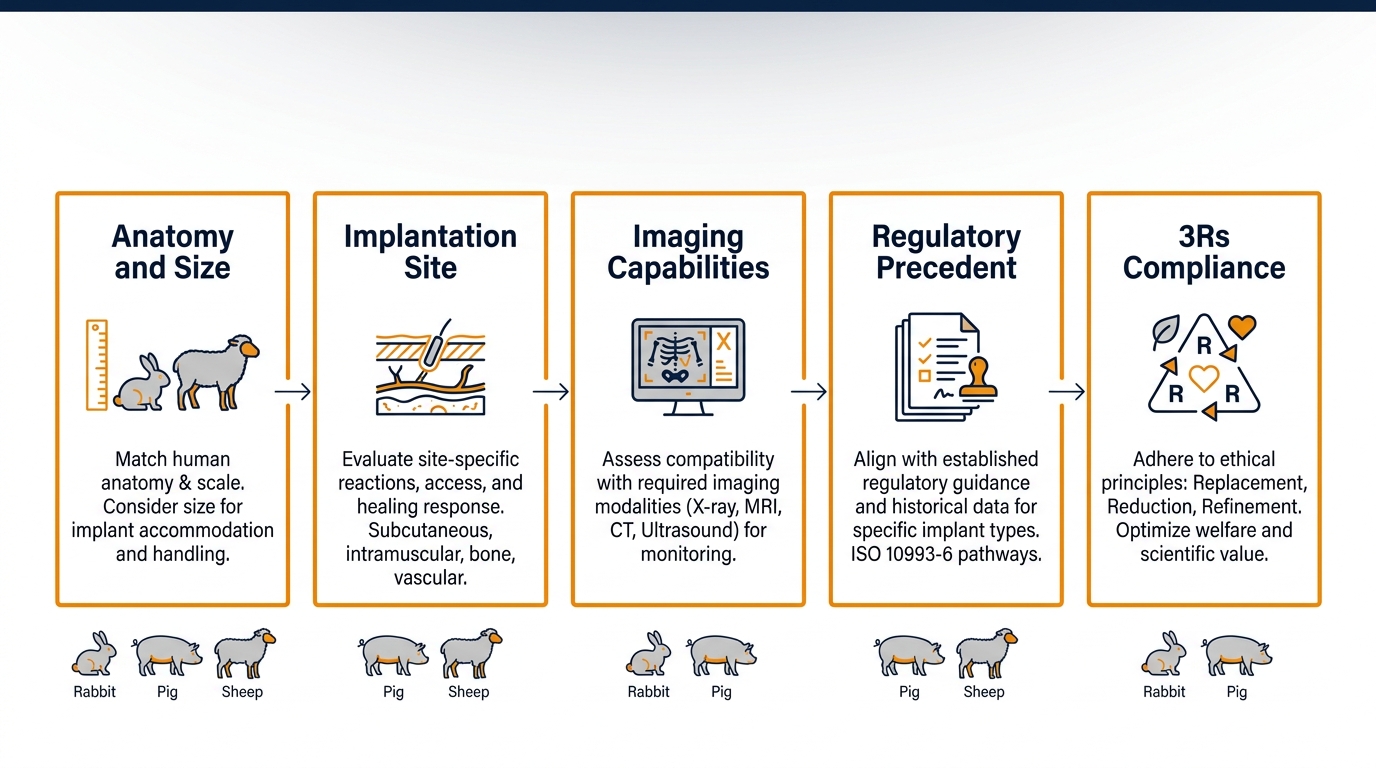
Selecting the appropriate implant safety study animals is a key step that influences data quality, regulatory acceptance, and translational value. Selection is based on anatomy, implant size, implantation site, expected mechanical loading, and the ability to measure relevant endpoints.
Small animal models — mice, rats, rabbits — are well suited for initial screening, subcutaneous local-effect testing, and material-level biocompatibility per ISO 10993-6. Large animal models such as pigs, sheep, and goats are used when the study must mimic clinical procedures, accommodate human-scale devices, or replicate cardiovascular, orthopedic, or ophthalmic anatomy. Large animal facilities also enable use of clinical-grade imaging, fluoroscopy, echocardiography, and laparoscopic platforms, which strengthens translational relevance and supports surgeon training on the actual device.
Key Criteria for Animal Model Selection
- Implantation site suitability: subcutaneous, intramuscular, bone, vascular, neural
- Imaging and clinical measurement capabilities available in-house
- Anatomical and mechanical compatibility with the device design
- Regulatory precedent for the indication and target market
- Ethical considerations under the 3 Rs framework (Replace, Reduce, Refine)
- Rabbit models are widely used for local effects after implantation due to well-characterized tissue response patterns
The Role of ISO 10993-6 in Implantation Testing
A fundamental aspect of implant preclinical evaluation is adherence to standards like ISO 10993-6, which focuses on evaluating local effects after implantation around a medical device or material. The standard prescribes implantation sites, control articles, observation periods, and a histological scoring framework covering inflammation, necrosis, fibrosis, neovascularization, fatty infiltrate, and giant cell presence.
Histopathology under ISO 10993-6 produces a semi-quantitative comparison between test and control sites, yielding an irritation or reactivity index that regulators can interpret consistently across submissions. The standard’s value lies in this standardized, comparable language for tissue response — turning subjective observation into structured evidence.
ISO 10993-6 addresses local tissue effects only. It does not fully cover systemic toxicity, genotoxicity, carcinogenicity, immunogenicity, or device-specific functional performance. A complete biological evaluation combines 10993-6 with other parts of the ISO 10993 series and, when relevant, functional and pharmacokinetic studies.
Essential Endpoints for Implant Safety Studies
Endpoint selection for a chronic implant study integrates clinical, laboratory, imaging, and histopathological data to build a layered safety picture supporting long-term implant safety conclusions.
Clinical Monitoring
Body weight, behavior, food intake, signs of pain or infection, and general health throughout the study.
Hematology & Chemistry
Systemic toxicity screening through blood panels detecting organ stress, immune activation, or leachable accumulation.
In Vivo Imaging
Radiography, ultrasound, CT, MRI, and OCT assess migration, integration, patency, and structural integrity without terminal sacrifice.
Histopathology
Microscopic examination of explanted tissue for inflammation, fibrous capsule thickness, necrosis, and foreign body reaction — the cornerstone of ISO 10993-6.
Why is Histology Considered the Most Robust Evidence?
Histology delivers direct, microscopic evidence of the biological response at the tissue–implant interface, allowing controlled comparison against negative or predicate controls. It is the only modality that visualizes cellular populations, capsule architecture, and material–tissue boundaries simultaneously — which is why it remains the cornerstone of ISO 10993-6 evaluations. The FDA maintains a structured mapping of recommended endpoints by device category, providing a useful starting point for protocol design.
Mapping Business Needs to Preclinical Capabilities
Sponsors approaching implant studies often share similar operational needs but face different scientific challenges. The table below maps common needs to practical capabilities a well-equipped preclinical partner provides.
| Business Need | How an Integrated Facility Supports It |
|---|---|
| Surgical realism for human-scale devices | Large animal surgery rooms with C-Arm fluoroscopy, echocardiography, and 4K laparoscopy |
| Regulatory-grade documentation | GLP-aligned procedures, structured reports, full traceability of test article and data |
| Reduced timelines | In-house animal housing, surgery, imaging, and pathology coordination under one roof |
| Ethical compliance under Israeli law | IACUC-equivalent oversight and 3Rs-based protocol design per Animal Welfare Law, 1994 |
| Cross-disciplinary protocols | Senior surgeons and scientific escort across cardiology, orthopedics, and ophthalmology |
Designing Regulatory-Compliant Implantable Device Testing
A risk-based approach is the foundation of credible implantable device testing. Protocol design begins with intended use, contact category and duration, patient population, and a documented risk analysis that defines which endpoints are necessary and which can be justified out.
Critical design elements include implantation sites and quantities per timepoint, inclusion/exclusion criteria, randomization and blinding where feasible, control articles, statistical analysis plans, and reporting templates aligned with regulatory expectations. Our team assists clients in designing implant preclinical evaluation protocols that stand up to the most rigorous regulatory scrutiny.
GLP: When Is It Mandatory, and When Is It an Advantage?
Good Laboratory Practice is typically required for definitive safety studies that directly support regulatory submissions to the FDA, EMA, and other authorities. Even when not strictly mandatory — for example, in early feasibility work — GLP-aligned conduct provides quality, integrity, and traceability that strengthen the data package and significantly reduce the risk of deficiency letters and rework cycles.
Determining Animal Numbers for Chronic Implant Studies
No fixed number applies across studies. Animal numbers depend on statistical needs, number of timepoints, expected variability, control groups, and the sensitivity of primary endpoints. The 3 Rs principle — Replacement, Reduction, Refinement — guides the design toward the minimum number compatible with scientific validity.
Repeated, non-terminal measurements such as imaging or blood sampling can reduce overall numbers by allowing each animal to serve as its own longitudinal control. In Israel, animal use in research is regulated under the Animal Welfare (Experiments on Animals) Law, 1994, and overseen by the Council for Animal Experimentation under the Ministry of Health — a framework that aligns naturally with international 3Rs expectations.
Comparing Acute vs. Chronic Implant Study Designs
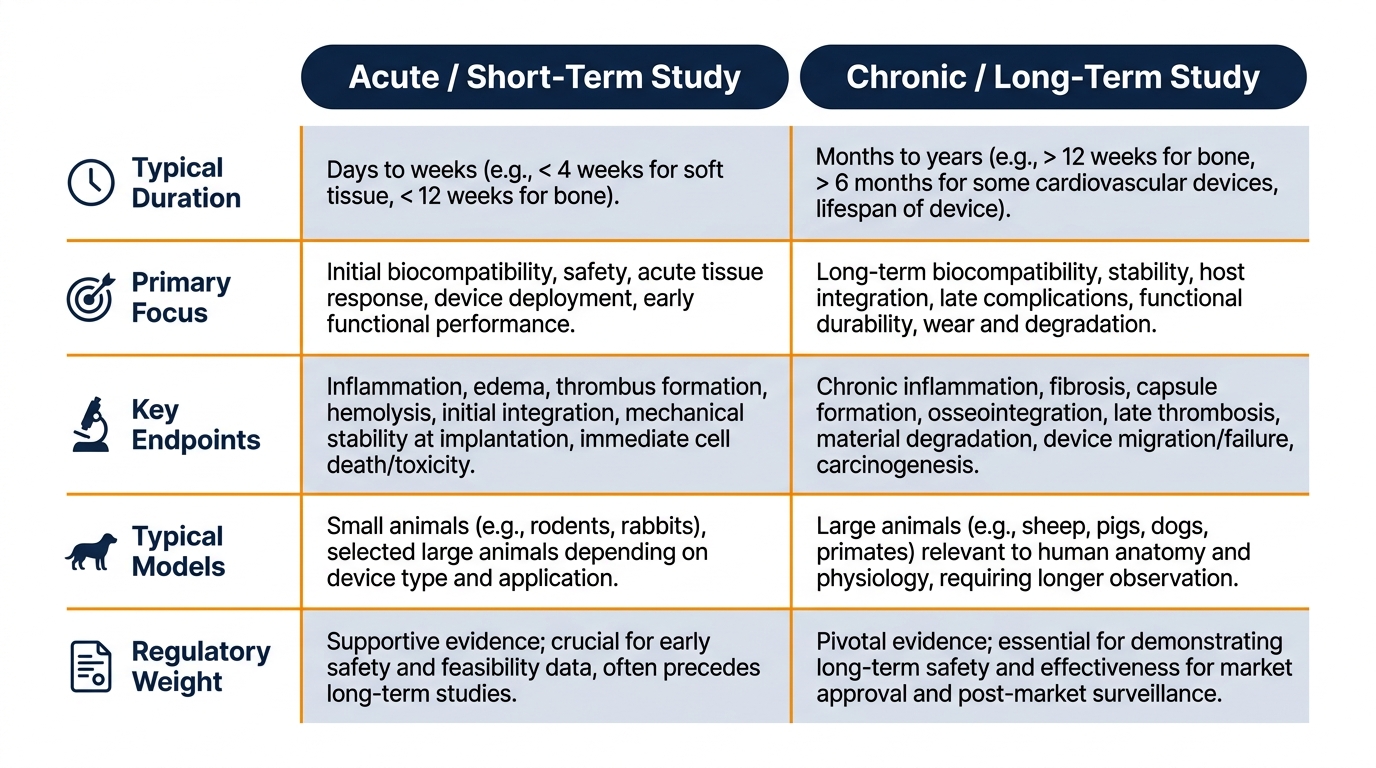
| Parameter | Acute / Short-Term Study | Chronic / Long-Term Study |
|---|---|---|
| Typical duration | 1–28 days | 13 weeks to 12+ months |
| Primary focus | Surgical feasibility, deployment, acute response | Tissue remodeling, durability, late events |
| Key endpoints | Procedure success, acute inflammation markers | Capsule maturation, degradation, mechanical integrity |
| Typical models | Rodents, rabbits, pigs (acute non-survival) | Sheep, pigs, goats (long-term survival) |
| Regulatory weight | Supportive / feasibility level | Pivotal for permanent implants |
What Can Potentially Replace Animal Testing for Implants?
Global momentum behind reducing and replacing animal testing has produced advanced in vitro models — organ-on-chip platforms, 3D co-cultures, perfused tissue constructs — and increasingly sophisticated computational modeling, including finite element analysis and physiologically based pharmacokinetic simulations.
These tools are valuable for screening, mechanism-of-action work, and refining hypotheses before in vivo studies. However, they generally cannot fully replace animal studies for complex long-term implant safety evaluations, particularly where mechanical loading, multi-tissue interaction, immune dynamics, and chronic remodeling must be observed together.
Use alternatives where they shorten development and reduce animal use, and reserve in vivo studies for questions only living systems can answer. Our expertise extends to a range of pre-clinical toxicology studies, where alternative methods are explored to optimize testing strategies and refine animal-based protocols whenever scientifically justified.
Common Mistakes That Delay Regulatory Acceptance
Several recurring issues turn otherwise solid programs into rework cycles. Each of the following is avoidable with disciplined protocol design and an experienced scientific escort throughout the study lifecycle.
Unjustified Model Selection
Choosing an animal model without documented justification — especially when anatomy or biomechanics differ meaningfully from the human application — is a frequent regulatory finding.
Underestimating Timepoints
Underestimating timepoint requirements for permanent implants — running a 90-day study with no rationale for omitting longer endpoints — generates predictable deficiency letters.
Missing Control Articles
Omitting appropriate negative or predicate controls makes histopathological interpretation unreliable and eliminates the comparative reference regulators expect.
Unstructured Histopathology
Providing histopathology without a scoring framework consistent with ISO 10993-6 produces subjective, non-comparable data that undermines the submission’s credibility.
Poor Traceability Chain
Insufficient documentation of the test article’s identity, sterilization, and handling chain can call the entire study’s integrity into question — even when the science is otherwise sound.
“Regulatory deficiencies in preclinical implant studies are rarely scientific failures — they are documentation and design failures. A well-structured protocol, properly executed and fully traceable, is the single most reliable path to on-time regulatory acceptance.”
— Adir Koreh, CEO, BIOTECH FARM Ltd.
Frequently Asked Questions
Ready to Advance Your Implant Program?
What does your device need to demonstrate before it reaches the first patient — and is your current preclinical plan structured to deliver that evidence efficiently? Bringing an implantable medical device to market demands scientific precision, regulatory fluency, ethical responsibility, and a partner equipped to manage every phase under one roof.





