Medical Device Preclinical Regulatory Pathway: 510(k), De Novo & PMA Explained
Bringing a medical device from concept to clinical use is rarely a straight line. The medical device preclinical regulatory pathway is the structured sequence of laboratory, bench, and in some cases animal investigations that generate the evidence regulators need to authorize a device for human use. It is the bridge between an engineering prototype and a marketable product, translating design intent into documented safety and performance.
For manufacturers, early planning of this pathway is not optional. The risk classification of the device, the intended clinical claims, and the target jurisdictions all shape what tests are required, in what order, and under what quality system. Treating preclinical strategy as an afterthought typically leads to repeated studies, regulatory hold letters, and significant time-to-market delays — outcomes that careful upfront mapping can largely prevent.
Expert Insight from BIOTECH FARM Ltd.
The most costly preclinical failures we observe are not scientific failures — they are planning failures. Developers who integrate regulatory strategy into design controls from day one consistently reach submission faster and with fewer surprises. Identifying your target pathway, predicate, and key study endpoints before a single prototype is tested is the single highest-return investment in any medical device program.
Understanding Regulatory Pathways for Medical Devices in the US: 510(k), De Novo, and PMA
In the United States, the FDA assigns medical devices into Class I, II, or III based on the level of risk they pose to patients and users. This classification is the single most important factor in determining which submission pathway applies. Low-risk devices may be exempt from premarket review altogether; moderate-risk devices typically follow 510(k) or De Novo; and high-risk devices generally require Pre-Market Approval (PMA).
Choosing among 510(k), De Novo, and PMA is not only about risk class. It also depends on whether a comparable legally marketed device already exists, the novelty of the technology, and the strength of available scientific evidence. A clear regulatory strategy aligns the device’s risk profile with the most efficient pathway that still satisfies the agency’s evidentiary expectations.
What is the 510(k) pathway and what makes it unique?
The 510(k), or Premarket Notification, is built around the concept of Substantial Equivalence. A manufacturer demonstrates that the new device is as safe and effective as a legally marketed “predicate device” with the same intended use and either the same technological characteristics or different ones that do not raise new questions of safety and effectiveness. Identifying an appropriate predicate — and rigorously comparing indications, design, materials, and performance — is therefore the first analytical step.
The 510(k) is generally appropriate for Class II devices and some Class I devices not exempt from notification. Even within this pathway, robust preclinical testing for medical devices is typically expected to support the equivalence argument.
De Novo Classification: When Innovation Meets Lower Risk
The De Novo pathway is designed for novel devices that are low-to-moderate risk but have no suitable predicate. Instead of forcing such devices into the much more burdensome PMA process by default, De Novo allows the FDA to classify them into Class I or Class II with appropriate special controls. Compared to 510(k), De Novo does not rely on equivalence; compared to PMA, it does not require the same depth of independent clinical evidence. The result is a proportionate route that rewards genuine innovation without overburdening developers of moderate-risk technologies.
Pre-Market Approval (PMA): The Most Rigorous Pathway for High-Risk Devices
PMA is the most stringent FDA pathway and is generally reserved for Class III devices — those that support or sustain human life, are of substantial importance in preventing impairment of health, or present a potential unreasonable risk of illness or injury. PMA requires independent scientific evidence providing a reasonable assurance of safety and effectiveness.
- Nonclinical bench data and biocompatibility evaluation
- Sterilization validation where relevant
- Software and electrical safety where applicable
- Animal studies where justified
- Well-controlled clinical investigations
Deeper Dive into Preclinical Requirements per Pathway
While the three US pathways share a common scientific vocabulary, the specific preclinical expectations differ significantly. Understanding these differences helps developers scope studies appropriately and avoid both under-testing and over-testing.
510(k) Preclinical Requirements
Center on performance (bench) testing tailored to intended use and comparison with the predicate. Covers biocompatibility (ISO 10993), sterilization, software V&V, and EMC/electrical safety. Animal studies are not a default requirement — they are considered when bench testing alone cannot characterize performance.
De Novo Preclinical Data
Without a predicate to anchor equivalence, manufacturers must comprehensively characterize the device, identify all reasonably foreseeable risks, and demonstrate that mitigations adequately address them. Bench, biocompatibility, software, sterilization, and human factors evaluations are commonly bundled.
PMA Preclinical Requirements
Substantially more demanding. Because PMA requires independent evidence of safety and effectiveness, studies must be extensive, rigorously documented, and linked to device claims. Preclinical data inform the clinical investigation design, justify first-in-human exposure, and support the IDE. Animal testing is most often required for implants, tissue-interacting devices, or novel mechanisms.
Does 510(k) Preclinical Testing Always Require Animal Studies?
No. Animal studies are not a default 510(k) requirement. They are considered when bench and analytical testing alone cannot adequately characterize device performance — for example, when there are meaningful technological differences from the predicate, complex tissue interactions are at play, or biological response to a new material cannot be fully predicted in vitro.
The FDA actively encourages use of non-animal methods when scientifically valid, in line with broader efforts to apply the 3Rs principles to nonclinical research. Well-designed programs maximize bench and in vitro methods before escalating to in vivo models, ensuring every animal study is scientifically justified and properly powered.
Does the De Novo Pathway Always Require Clinical Data?
Not always. If preclinical evidence — bench, analytical, biocompatibility, and usability — convincingly demonstrates safety and effectiveness for a moderate-risk indication, clinical data may not be required. In other cases, targeted clinical information is needed to confirm performance in the intended use environment. The decision is risk-based and tied to how completely preclinical models reflect actual clinical conditions.
A Quick Comparison: 510(k) vs. De Novo vs. PMA
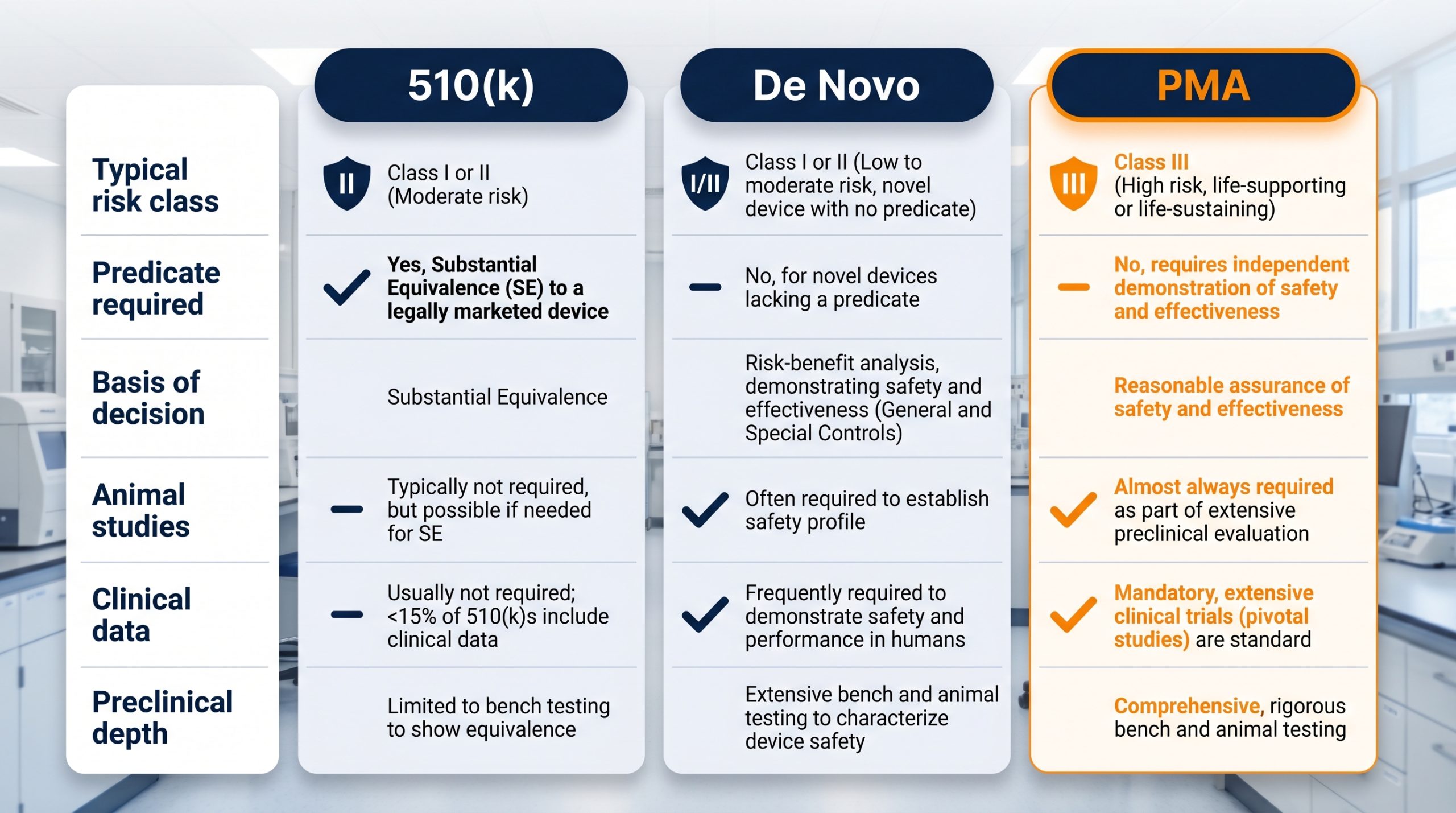
| Aspect | 510(k) | De Novo | PMA |
|---|---|---|---|
| Typical Risk Class | Class II (some Class I) | Class I or II, novel | Class III |
| Predicate Required | Yes | No | No |
| Basis of Decision | Substantial Equivalence | Risk-based classification with special controls | Independent safety & effectiveness evidence |
| Animal Studies | Sometimes | Sometimes | Frequently |
| Clinical Data | Rarely | Sometimes | Almost always |
| Preclinical Depth | Focused on equivalence | Comprehensive, risk-based | Most extensive |
Essential Components of Preclinical Testing
Regardless of pathway, preclinical evaluation rests on a set of recurring building blocks. The mix and depth vary, but the underlying logic — characterize the device, anticipate failure modes, and verify performance against predefined criteria — is universal.
Bench Testing
In-vitro performance and functional evaluations on device components. Covers functional performance, mechanical durability, dimensional accuracy, simulated use, and worst-case scenario testing. Defining “worst-case” rigorously gives bench testing its predictive value.
Biocompatibility (ISO 10993)
Evaluates the device’s ability to perform without unacceptable biological response. Addresses cytotoxicity, sensitization, irritation, systemic toxicity, genotoxicity, and implantation effects. FDA guidance frames this as a risk-based exercise starting with chemical and physical characterization.
Sterilization Validation
For sterile-labeled devices, validation is non-negotiable. Establishes that the chosen method — EO, gamma, e-beam, steam — reliably achieves the required Sterility Assurance Level (SAL) of 10⁻⁶. Documentation must address product/packaging compatibility, bioburden, and revalidation triggers.
Software Validation (SaMD)
Devices with software — SaMD or embedded firmware — require rigorous verification and validation. Covers functional testing, cybersecurity controls, reliability under stressed conditions, and usability for intended users. FDA guidance specifies documentation scaled to the software’s level of concern.
Electrical Safety & EMC
Electrically powered devices must satisfy the IEC 60601 series. EMC testing verifies correct function in the intended electromagnetic environment and absence of undue interference with other equipment. Essential for real-world reliability in hospitals and clinics as well as regulatory acceptance.
“Treating documentation as a deliverable from day one — rather than a write-up at the end — is one of the most effective countermeasures against regulatory delay.”
— BIOTECH FARM Ltd. Regulatory Team
The Role of GLP in Preclinical Studies

Good Laboratory Practice (GLP) is the quality system that governs the conduct of nonclinical health and environmental safety studies. In the US, the foundational regulation is 21 CFR Part 58, which addresses organization, personnel, facilities, equipment, standard operating procedures, study protocols, study conduct, records, and reports.
GLP compliance is typically required for nonclinical safety studies that support regulatory submissions such as IDEs and PMAs, where data integrity directly underpins the agency’s confidence in the evidence. Performance bench tests and many engineering evaluations are not necessarily GLP, but pivotal safety studies in animals usually are.
For developers, the practical implication is that the GLP decision must be made early: retrofitting GLP onto a study already in progress is rarely successful. A clear list of which studies will be GLP, conducted at qualified facilities with documented quality systems, protects the entire submission from avoidable challenges.
21 CFR Part 58: What GLP Covers
- Organization and personnel qualifications
- Facilities and equipment requirements
- Standard operating procedures (SOPs)
- Study protocols and conduct documentation
- Records, archives, and final reports
Preclinical Data for CE Marking: Navigating the EU MDR
For market entry in the European Union, the Medical Device Regulation (Regulation (EU) 2017/745, MDR) governs what manufacturers must demonstrate. CE mark preclinical data are integrated into the Technical Documentation that supports the conformity assessment. The MDR places strong emphasis on a continuous risk management system, a thorough clinical evaluation, and active Post-Market Surveillance (PMS) — and each of these pillars relies on robust preclinical foundations.
Annex II defines the structure of the Technical Documentation, while Annex XIV details the clinical evaluation process. Together, they make preclinical evidence inseparable from the European route to market.
What is Included in the Technical File for CE Marking?
Device Description & Specification
Complete description of the device, its intended purpose, patient population, indications, contraindications, and technical specifications.
GSPRs & Risk Management
General Safety and Performance Requirements demonstrating compliance, alongside comprehensive benefit-risk analysis and risk management documentation per ISO 14971.
Verification & Validation
Bench performance, biocompatibility, sterilization, software, electrical safety, EMC, stability, shelf life, and animal study reports when relevant — all explicitly linked to identified risks.
Mapping Business Needs to Preclinical Support
| Business Need | How an Experienced Preclinical Partner Supports It |
|---|---|
| Early go/no-go decisions | Pilot studies and feasibility models that quickly expose design weaknesses before major investment |
| Regulatory-grade safety data | GLP-aligned study conduct with well-documented procedures and traceable records |
| Complex implant or interventional testing | Surgical capabilities, imaging support, and large animal models suited to cardiology, orthopedics, ophthalmology, and more |
| Tailored study design | Protocols matched to the device’s intended use, claims, and target submission rather than a one-size-fits-all template |
| Integration across disciplines | Coordinated bench, biological, and in-vivo work under a single project plan to reduce handoffs and timeline gaps |
| Ethical and transparent execution | Animal welfare practices and transparent reporting aligned with the 3Rs principles |
Common Pitfalls and Best Practices in Preclinical Planning
Most preclinical setbacks are not scientific surprises — they are planning failures. Developers who treat preclinical strategy as a continuation of design controls, rather than as an isolated testing phase, consistently move faster through regulatory review.
How Early Should a Pre-Submission Meeting with the FDA Be Considered?
For novel devices, devices on the boundary between pathways, or programs with unusual preclinical questions, a Pre-Submission (Pre-Sub) meeting should be considered as soon as the testing strategy is sketched but before pivotal studies start. Early engagement clarifies regulator expectations about test methods, acceptance criteria, animal models, and clinical endpoints. The investment of time in preparing a Pre-Sub briefing package is usually returned many times over in avoided rework.
Common Causes of Regulatory Delays
- Absence of clearly defined acceptance criteria before testing begins
- Insufficient or non-representative sample sizes
- Inadequate justification for chosen test methods
- Weak traceability between risks, design inputs, test plans, and results
- Disorganized reporting or undocumented protocol deviations
Best Practices for Preclinical Success
- Define acceptance criteria in the protocol before any testing begins
- Hold a Pre-Sub meeting early for novel or boundary-pathway devices
- Integrate preclinical strategy into design controls from the outset
- Treat documentation as a deliverable from day one
- Decide GLP status before studies commence — not mid-stream
Frequently Asked Questions
Strategic Preclinical Planning for Market Success
A thoughtfully designed medical device preclinical regulatory pathway is far more than a regulatory obligation — it is a foundation for clinical credibility, manufacturing maturity, and durable market access. The strongest programs treat preclinical work as an integrated scientific narrative that links risk analysis, design controls, bench evidence, biological evaluation, and, when needed, in-vivo studies into one coherent story for reviewers.
“The developers who reach clearance fastest are not those who test the most — they are those who test the right things, at the right time, with the right documentation, guided by a clear regulatory strategy from the very beginning.”
— Adir Koreh, CEO, BIOTECH FARM Ltd.
Are you mapping out the preclinical strategy for your next medical device and want experienced partners to help you design studies that match both your scientific questions and your target submission? Connect with BIOTECH FARM Ltd. to discuss how a tailored preclinical research and development approach can support your path from concept to clearance.





