Preclinical Protocol Writing Services
Audit-Ready Documents That Translate Science Into Executable Studies
For biotech, medical device, and pharmaceutical teams approaching IND-enabling work, the gap between a scientific concept and a study that actually runs — reproducibly, defensibly, and on time — often lies in one document: the protocol. Our preclinical protocol writing services are built for sponsors who need a clear, consistent, audit-ready plan that reflects regulatory expectations and operational reality.
Expert Insight
The most costly errors in preclinical programs rarely happen at the bench — they happen in the protocol document, weeks before the first animal enters study. A single ambiguous time point, an immeasurable humane endpoint, or a mismatched dosing table can invalidate an entire PK curve or render pathology data undefendable during an IND submission review. Audit-ready protocol writing is not a formality — it is your first line of scientific defence.
What Do Preclinical Protocol Writing Services Actually Include?
Direct answer: Preclinical protocol writing services involve gathering requirements, developing a written and executable study plan through structured preclinical study design consulting, defining endpoints, methods, basic statistics and analysis, the workflow for approvals, and producing a consistent document ready for implementation and quality review.
In practice, nonclinical protocol development spans a synopsis, schedule of activities, procedures, animal model details, dosing and administration, sampling timelines, bioanalysis plan, and deviation handling. The deliverable also reflects GLP versus non-GLP expectations: signatures, QA review, version control, and archiving for GLP; lighter formalism but the same scientific rigor for non-GLP feasibility work.
Common Deliverables
- Study synopsis and experimental design
- Test article and vehicle handling procedures
- Dosing tables and in-life schedule
- Sampling and bioanalysis annexes
- Pathology and necropsy plan
- Deviation and amendment framework with traceable signatures
What Clients Need at Kickoff
- Target product profile and prior in vitro/in vivo data
- Candidate model rationale and formulation details
- Regulatory strategy (GLP vs. exploratory)
- Preferred bioanalytical assays
- Timeline constraints and regulatory milestones
Why a Preclinical Study Protocol Decides Whether Your Data Will Be Usable
Direct answer: A preclinical study protocol defines “what, how, and why” before any animal enters study, and when written correctly it minimizes errors, reduces amendments, and shortens the time to actionable data.
“A good preclinical study protocol template closes the gaps between scientific intent, operational execution, and QA expectations — turning abstract objectives into measurable, time-stamped actions that any qualified technician can repeat without interpretation.”
— BIOTECH FARM Ltd. Protocol Development Team
Strong nonclinical protocol development also defines go/no-go criteria upfront — so when interim data arrive, the team is not negotiating the rules of success mid-study. It also defines go/no-go criteria upfront so when interim data arrive, the team is not negotiating the rules of success mid-study.
GLP Protocol Writing vs. Non-GLP Protocol Development: Where the Line Sits
Direct answer: GLP protocol writing requires stricter documentation, quality control, traceability, and signatures; non-GLP is more flexible but must still be clear and executable to produce reliable data.
When to Choose GLP
Pivotal safety studies supporting IND/CTA submissions almost always require GLP. If the data may later be cited in a regulatory dossier, default to GLP-grade documentation from day one.
GLP includes:
Formal version control · QA review · Defined roles · Deviation handling · Structured archiving
When Non-GLP Is Appropriate
Early proof-of-concept, pilot tolerability, model characterization, and method development typically run non-GLP. The Regulatory Compliance Preclinical Research resource maps GLP expectations against practical study realities.
Warning: Retrofitting GLP
Missing chain-of-custody records, undocumented deviations, and uncalibrated equipment mean data cannot be salvaged for pivotal use.
What Should a GLP Protocol Include to Pass QA Without Endless Revisions?

Direct answer: A robust GLP protocol writing deliverable includes precise objectives, a complete experimental design, measurement methods, deviation management, team roles, and appendices referencing SOPs — in a consistent format that supports auditing.
Typical breaking points include immeasurable endpoint definitions, open-ended inclusion/exclusion criteria, inconsistent sampling windows, and ambiguous dosing units. The OECD Principles on Good Laboratory Practice remain the global reference for documentation discipline.
✓ Pre-QA Internal Checklist
- Objectives are measurable and time-bound
- Endpoints map to specific methods and assays
- Doses, volumes, and routes are consistent across tables and text
- Humane endpoints are quantitative with defined decision authority
- All SOPs cited are current and version-controlled
- Signatures, dates, and version numbers are complete
⚠ Dangerous Language That Generates Deviations
Replace vague phrases with specific values:
- “As appropriate” → “every 24 ± 2 hours”
- “If needed” → “if body weight loss ≥ 20% from baseline”
- “Periodically” → “within 30 minutes of the scheduled time point”
- “At the discretion of the investigator” → [specific threshold + decision tree]
From Scientific Brief to Final Version: The Nonclinical Protocol Development Process
Direct answer: A disciplined nonclinical protocol development process starts with a scientific brief, proceeds to an outline plus clarification questions, then a draft 0.1, a structured feedback round, QA/compliance review, and a “final” with clear version management.
Process Stages at a Glance
→
2. Outline + Questions
→
3. Draft 0.1
→
4. Feedback Round
→
5. QA Review
→
6. Final + Version Lock
What We Need at Kickoff
- Candidate’s IB or data summary
- Prior tox/PK data and target indication
- Available CMC and formulation stability data
- Named decision-makers with authority to lock assumptions
Preventing Feedback Chaos
- Use a single tracked document with one editorial owner
- Consolidated comment matrix for all teams
- Run review cycles in defined time windows
- Record every closed decision in a versioned issue log
How Long Does It Take to Prepare a Preclinical Protocol — and What Causes Delays?
Direct answer: A well-scoped protocol typically takes between a few days and several weeks, depending on complexity, data availability, number of stakeholders, and QA/approval rounds.
Common Delay Factors
- Unclosed endpoints and indecision on animal model
- Late dose or group structure changes
- Limited availability of bioanalysis or histopathology vendors
- Multiple review stakeholders without a clear decision owner
Most Reliable Acceleration Tactic
A short pre-writing decision document — two to three pages capturing locked assumptions — before the team commits to a full draft. This is where strong preclinical protocol writing services earn their value: by forcing decisions before they become amendments.
Animal Study Protocol Preparation: Writing What Is Actually Executable
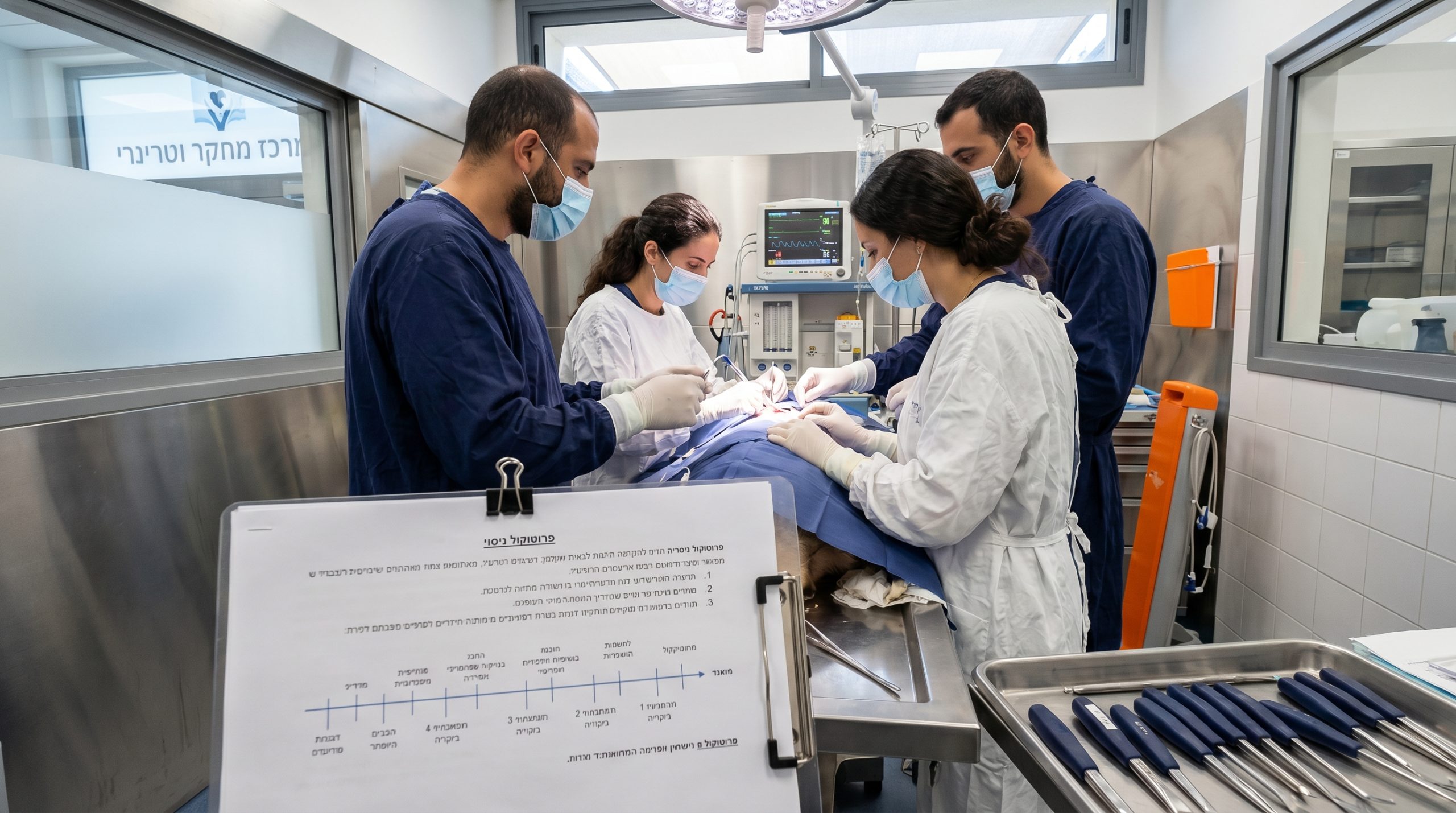
Direct answer: Animal study protocol preparation breaks the experiment into measurable actions — model selection, randomization, dosages, welfare monitoring, sampling points, and humane definitions — so every team executes without interpretation.
In Israel, animal work is governed by חוק צער בעלי חיים (ניסויים בבעלי חיים), התשנ״ד‑1994, and protocol authors must align with institutional approval requirements as well. Practical context on welfare-compliant execution can be found in our overview of GLP Animal Studies.
Reducing Rework Risk During Execution
- Lock randomization scheme and define exclusion criteria quantitatively
- Pre-specify replacement animal rules
- Dosing volumes account for species weight ranges
- Pre-print all in-life forms verified against protocol schedule verbatim
Defining Humane Endpoints Measurably
Replace narrative descriptions with quantitative thresholds:
- Percent body weight loss from baseline
- Body condition score with defined scale and threshold
- Respiratory rate ranges or specific clinical signs persisting over defined interval
- Decision authority and documentation requirements written directly into protocol
Do You Provide a Preclinical Study Protocol Template, and What Is Its Advantage?
Direct answer: Yes — a preclinical study protocol template shortens time, prevents missing sections, and creates uniformity across studies, which facilitates QA, knowledge transfer, and report writing.
Faster Drafting
Placeholders with guidance for each section — authors know what to write and why.
Cross-Study Consistency
Standardization across teams and sites — essential when a program runs parallel studies or expands to multiple species.
Shorter QA Cycles
Reviewers know exactly where to look — reducing review time and new-team onboarding costs.
Aligning the Protocol with SOPs, Forms, and GLP Documentation
Direct answer: Alignment is achieved through cross-references to SOPs, definition of required forms and logs, and early harmonization of the protocol, raw data expectations, and reporting plan.
The Traceability Mapping Flow
→
SOP
→
Form
→
Data
→
Report
Regulatory references such as the FDA’s Records and Reports (GLP) guidance reinforce why this traceability matters. Each deviation has a defined route through QA, and the traceability map is maintained as a living document from draft through archiving.
Which Protocol Sections Cause the Most Amendments — and How to Minimize Them
Direct answer: Most amendments come from unclosed sections: design, endpoints, schedule, statistics/groups, and exclusion criteria. Closing each with a measurable decision before “final” dramatically reduces amendment frequency.
“The discipline behind effective preclinical protocol writing services is the relentless removal of interpretive words. Maintain an assumption list alongside the draft, and a decision log that records who agreed to what, when, and on what basis.”
— BIOTECH FARM Ltd. Protocol Writing Methodology
Choosing Endpoints, Groups, and Animal Numbers to Serve Both Science and Regulation
Direct answer: Select endpoints that directly measure the hypothesis, define statistical power to justify animal numbers, and respect regulatory expectations for group sizes and study duration. Apply the 3Rs principle (Replacement, Reduction, Refinement) consistently, and consult biostatisticians early rather than after the design is “final.”
In Israel, group composition and welfare considerations are framed by תקנון המועצה לניסויים בבעלי חיים. For format-level consistency, reference standards such as the common templates for nonclinical studies developed by industry consortia help align expectations between sponsors, CROs, and reviewers.
Comparison Table: GLP vs. Non-GLP Protocol Expectations
| Element | GLP Protocol | Non-GLP Protocol |
|---|---|---|
| Typical Use | Pivotal safety, IND-enabling studies | Feasibility, dose ranging, model development |
| QA Oversight | Mandatory, formal QA unit | Optional, internal review |
| Signatures | Study Director, Sponsor, QA | Principal investigator, optional sponsor |
| Version Control | Strict, archived | Recommended, often informal |
| Deviation Handling | Documented, signed, QA-reviewed | Logged, less formal |
| Raw Data Archiving | Long-term, defined retention | Project-dependent |
| Regulatory Acceptance | Submission-grade | Supportive only |
Business Needs Mapped to How Our Process Helps in Practice
| Business Need | How the Process Helps |
|---|---|
| Faster time from concept to in-life | Pre-writing decision document locks assumptions and removes mid-draft rework |
| Reduced amendments after approval | Quantitative endpoints, measurable humane criteria, and SOP cross-references built in from draft 0.1 |
| Audit-ready documentation | Version control, signed approvals, and traceability map maintained through final |
| Lower internal workload for sponsors | Single editorial owner, consolidated comment matrix, defined review windows |
| Smooth handoff between teams | Standardized template structure that bioanalysis, pathology, and operations recognize immediately |
| Alignment with Israeli and international frameworks | Mapping to local approval requirements alongside OECD/ICH expectations |
Frequently Asked Questions
Ready to Move Your Protocol From Draft to Audit-Ready?
Turn Scientific Intent Into Defensible Data
Whether you are preparing your first IND-enabling study, aligning a multi-site program, or trying to recover a stalled protocol before the next review cycle, the right document discipline is what turns scientific intent into defensible data.





