The Preclinical Package for Biologics: What’s Essential for a Confident IND Submission
Translating a promising biologic candidate into a first-in-human (FIH) study demands a coherent, science-based preclinical package that satisfies regulators while protecting trial participants. Unlike small molecules, biologics carry unique scientific complexities: target specificity, immunogenicity potential, species relevance, and intricate bioanalytical demands. This article unpacks what an IND-enabling package for biologics actually contains, how it is structured, and where development teams most often stumble. Drawing on ICH guidance (S6(R1), M3(R2), S7A, Q5C, Q6B) and over three decades of hands-on large-animal preclinical experience, we map the deliverables, decisions, and trade-offs that define a confident IND submission.
Expert Insight
The single most consequential decision in biologic preclinical development is not which assay to run — it is which species to use. A pharmacologically irrelevant species produces data that regulators cannot rely upon, regardless of GLP compliance. Species selection deserves the same strategic attention as clinical endpoint selection.
What Is a Preclinical Package for Biologics (IND-Enabling) and What Is Its Purpose?
A preclinical package for biologics is the comprehensive set of non-clinical studies, raw data, validated methods, and documentation that supports the safety rationale and scientific justification for FIH trials, ultimately enabling an Investigational New Drug (IND) or Clinical Trial Application (CTA) submission. Its primary goal is to demonstrate that the biologic drug development preclinical candidate is reasonably safe for human administration and has a credible mechanistic basis for further investigation.
A typical package addresses pharmacology and efficacy, GLP-compliant toxicology, safety pharmacology, pharmacokinetics (PK) and toxicokinetics (TK), immunogenicity, and the relevant Chemistry, Manufacturing, and Controls (CMC) attributes of the test article. Each element should connect to the proposed clinical protocol, dose, route, and patient population.
Why Biologics Demand a Different Playbook Than Small Molecules
The defining difference is the molecule itself. Biologics are large, target-selective, and often species-specific, so the biological relevance of the animal model dominates study design rather than a fixed testing battery. Immunogenicity assessment, ligand-binding bioanalysis, and exposure interpretation in the presence of anti-drug antibodies (ADAs) become central rather than peripheral.
Small molecule programs typically follow a more standardized matrix that includes genotoxicity, metabolism, and CYP-related interactions. For biologics, genotoxicity is generally not required, off-target effects are typically tied to exaggerated pharmacology rather than chemical reactivity, and species selection is driven by target binding rather than metabolic similarity.
Essential Components of Every Preclinical Package for Biologics Before IND
The core building blocks include relevant pharmacology and proof-of-concept studies, GLP repeat-dose toxicology in a relevant species, integrated PK/TK with validated bioanalysis, an immunogenicity strategy, and CMC documentation linking the manufactured material to the studied lots. Regulators expect a coherent narrative that ties activity, exposure, safety margin, and product quality into a defensible biologic IND preclinical rationale.
Typical Deliverables Often Overlooked in the Scope
Beyond the headline studies, programs frequently underestimate signed final reports, raw data traceability, validated analytical methods, the stability program, sampling and storage plans, and quality assurance audits. These deliverables determine whether the data can be used by regulators, not merely whether they exist.
???? Core Package Components
- Pharmacology & proof-of-concept
- GLP repeat-dose toxicology
- Integrated PK/TK with validated bioanalysis
- Immunogenicity strategy & ADA sampling
- CMC documentation & batch traceability
⚠️ Frequently Overlooked Deliverables
- Signed final GLP reports
- Raw data traceability records
- Validated analytical methods documentation
- Stability program data
- QA audits & deviations handling
Inside GLP Toxicology for Biologics
GLP toxicology for biologics centers on repeat-dose studies that mirror the proposed clinical regimen, integrating clinical observations, hematology, clinical chemistry, gross and histopathology, and TK measurements. The choice of a relevant animal species — one that expresses the target and responds to the biologic — is the single most consequential design decision. When no fully relevant species exists, scientific justification combined with surrogate molecules, transgenic or humanized models, or single-species programs may be defensible.
According to ICH S6(R1), nonclinical safety evaluation of biotechnology-derived pharmaceuticals should be science-based and flexible, with relevance of the test system as a guiding principle.
How Are Doses and NOAEL/Safety Margins Determined for Biologics?
Dose selection typically defines a high dose covering the maximum feasible exposure or a clear pharmacologic plateau, a low dose anchored near anticipated efficacy, and an intermediate dose to support exposure-response interpretation. The No Observed Adverse Effect Level (NOAEL), combined with TK exposure data, then informs a safe FIH starting dose, generally guided by ICH M3(R2) principles for timing and scope of nonclinical safety packages.
Scenario: When the Only Relevant Species Is Non-Human Primate
???? Case Study: mAb with NHP-Only Target Expression
Imagine a monoclonal antibody whose target is expressed only in non-human primates (NHP). A pragmatic path includes:
- Pilot PK/PD characterization in NHP
- GLP repeat-dose tox in a single relevant species with integrated safety pharmacology endpoints
- Comprehensive immunogenicity sampling throughout study
- Supporting in vitro tissue cross-reactivity data
The regulatory file then justifies single-species use through binding affinity comparisons, target distribution mapping, and pharmacology bridging.
Safety Pharmacology for Biologics: Is a “Core Battery” Always Necessary?
Safety pharmacology evaluates potential undesirable effects on vital organ systems — cardiovascular, respiratory, and central nervous system. ICH S7A defines the core battery, but for biologics these endpoints can frequently be integrated within repeat-dose toxicology studies through telemetry, respiratory plethysmography, and functional observation batteries.
Integration Advantage
Integration of safety pharmacology endpoints within repeat-dose toxicology studies reduces animal use, accelerates timelines, and aligns with the 3R principles when properly planned and quality-assured. This approach is well-accepted by regulators when the study design is sufficiently robust.
PK/TK for Biologics and What Must Be Measured
PK and TK studies must demonstrate systemic exposure across dose levels and time, characterize accumulation on repeat dosing, and establish exposure-toxicity and exposure-efficacy relationships. Bioanalysis for biologics relies on ligand-binding assays (ELISA, ECL), neutralization assays, and at times measurement of complexes, free vs. total drug, or downstream biomarkers. Validation per regulatory expectations is essential before pivotal samples are run.
Why ADA Can “Break” PK and How to Plan Around It
Anti-drug antibodies can accelerate clearance, mask exposure, or shift apparent half-life, undermining interpretation of toxicity findings. Sampling for binding and neutralizing ADAs at predefined toxicology and PK timepoints, paired with strategies to interpret reduced exposure, is essential. The EMA guideline on immunogenicity of therapeutic proteins details ADA assay strategies and the link between immunogenicity and PK/safety/efficacy.
ADA Risk Alert
Programs that do not pre-define ADA sampling timepoints and interpretive frameworks frequently find that immunogenicity findings invalidate exposure data — requiring costly repeat studies or weakening the regulatory submission. Build ADA strategy into study design from day one.
Immunogenicity Risk Assessment in the Preclinical Stage
Immunogenicity risk assessment is a structured evaluation of the likelihood and consequences of an immune response — including ADA generation, neutralizing antibodies, and cytokine release. It draws on sequence and structural analysis, aggregation and post-translational modification profiles, in vitro T-cell or MHC-binding assays where indicated, and a sampling strategy embedded into the toxicology and PK programs.
Outputs include a documented risk tier, a clinical monitoring plan, and assay readiness for future trials. For programs requiring specialized large molecule bioanalysis support, integrated capabilities in large molecule bioanalysis / immunogenicity / PK ensure that assay design is tightly coupled to study interpretation.
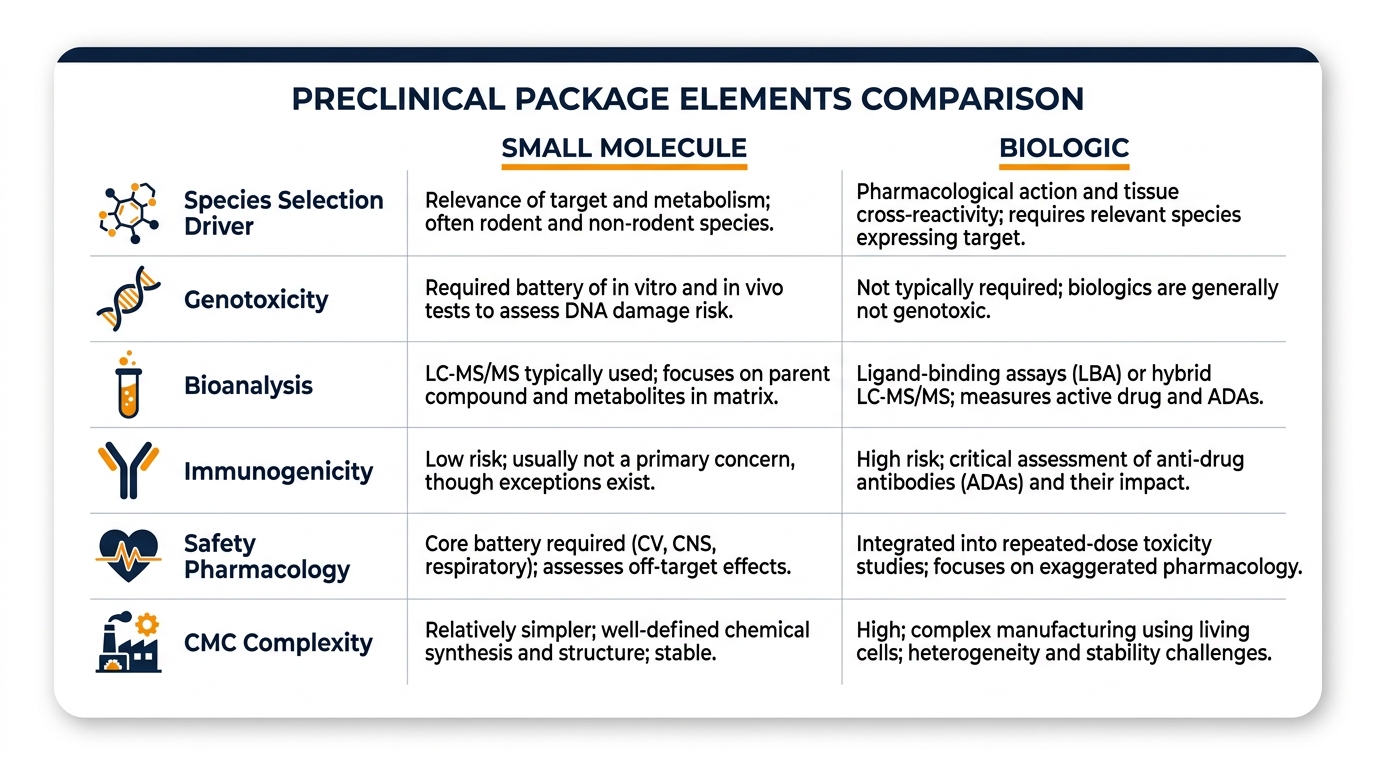
Comparison: Small Molecule vs. Biologic Preclinical Package
Understanding the structural differences between small molecule and biologic IND packages is essential for allocating resources appropriately and anticipating regulatory expectations.
| Element | Small Molecule | Biologic |
|---|---|---|
| Species selection driver | Metabolic similarity, ADME | Target binding & functional response |
| Genotoxicity | Standard battery (Ames, micronucleus) | Generally not required |
| Bioanalysis | LC-MS/MS | Ligand-binding, neutralization assays |
| Immunogenicity | Rarely a primary concern | Central to interpretation |
| Safety pharmacology | Often standalone core battery | Frequently integrated into tox |
| CMC complexity | Defined chemical entity | Process-defined product |
Do Biologics Require Genotoxicity Studies?
Standard proteins and monoclonal antibodies are not expected to interact directly with DNA, so traditional genotoxicity studies are typically not required. Exceptions arise for antibody-drug conjugates (ADCs), bioconjugates with cytotoxic payloads, novel linkers, or formulations containing reactive small-molecule excipients. In such cases, focused genotoxicity testing of the relevant component is appropriate.
For complex carrier systems, Drug Delivery Systems Preclinical Testing may include targeted genotoxicity work on excipients or carriers when justified by composition and route of administration.
Pharmacology and Efficacy Studies That Support a Biologic IND
Robust pharmacology demonstrates a clear mechanism of action, in vitro potency in the relevant biology, and in vivo confirmation in a model with reasonable translational value. The aim is not data volume but interpretive clarity: a credible dose-response, defensible biomarkers, and endpoints that map to the proposed clinical indication.
Selecting an In Vivo Model When a Perfect One Doesn’t Exist
When the human target is not adequately represented, options include surrogate molecules engineered for the test species, humanized or knock-in models, ex vivo human tissue systems, and biomarker-driven bridging strategies. Each option carries trade-offs in translational confidence and regulatory acceptance, and should be discussed with regulators early.
“The goal of preclinical pharmacology is not to predict clinical success with certainty, but to provide a credible mechanistic rationale that justifies exposing humans to the candidate — with a defined safety margin and an interpretable biomarker strategy.”
— Biotech Farm Scientific Team
Choosing a Relevant Species for Large Molecule Toxicology
Species selection rests on three pillars: functional binding to the target, a measurable and interpretable PK profile, and the feasibility of meaningful toxicity endpoints. When only one relevant species exists, robust justification supported by binding affinity data, tissue cross-reactivity, and pharmacological responsiveness is required.
The flexible, science-based framework articulated in ICH S6(R1) permits and even expects such tailored decisions for large molecule preclinical programs.
Swine Models
Humanoid organ size and anatomy make swine ideal for cardiovascular, surgical, and device-coupled biologic studies. Widely accepted for GLP programs.
Sheep & Goat Models
Valuable for orthopedic, pulmonary, and reproductive biologic programs where organ mass and physiology closely parallel human parameters.
Rabbit Models
Commonly used for ophthalmology, dermatology, and localized delivery biologic programs, with well-established regulatory precedent.
Test Article Characterization: The Gateway to Credible GLP Tox
Test article characterization is the documented evidence that the material dosed in animals has known identity, purity, potency, aggregation profile, and stability, with full batch traceability. Without it, even a flawlessly executed GLP toxicology study can be deemed regulatorily unusable — because the regulator cannot confirm what was actually administered.
Characterization data should bridge research-grade lots to GLP lots and ultimately to clinical material. This chain of evidence is non-negotiable and should be planned at program inception, not retrofitted after study initiation.
How CMC for “Preclinical” Differs from CMC for “IND”
Early preclinical work often uses material from less-refined processes, with tighter controls applied as the program matures. By IND, regulators expect detailed specifications, validated analytical methods, stability data per ICH Q5C, defined Critical Quality Attributes (CQAs), and robust batch documentation.
The narrative must show consistent manufacturing capability and a clear understanding of how product quality affects safety and activity. For programs partnering with full-scope preclinical facilities, the Biotech Farm approach combines large-animal capabilities, GLP study conduct, and scientific escort to align CMC realities with study design.
Specific Considerations for Biosimilar Preclinical Studies
The aim of biosimilar preclinical studies is to demonstrate similarity to a reference product, not to re-establish de novo safety. Programs emphasize comparative functional assays, comparative non-GLP PK/PD in animals, and often a focused comparative toxicology study only when residual uncertainty remains after extensive analytical comparability.
The WHO TRS 977 Annex 2 guidelines articulate this stepwise, totality-of-evidence approach for biosimilar development and regulatory assessment.
Comparability at IND
Comparability data may be needed to bridge material used in early preclinical studies to the clinical lot, ensuring that safety conclusions remain applicable. Analytical, biophysical, and where necessary nonclinical data form the comparability dossier — and must be established before IND submission, not after.
Realistic Timelines for IND-Enabling Biologic Studies
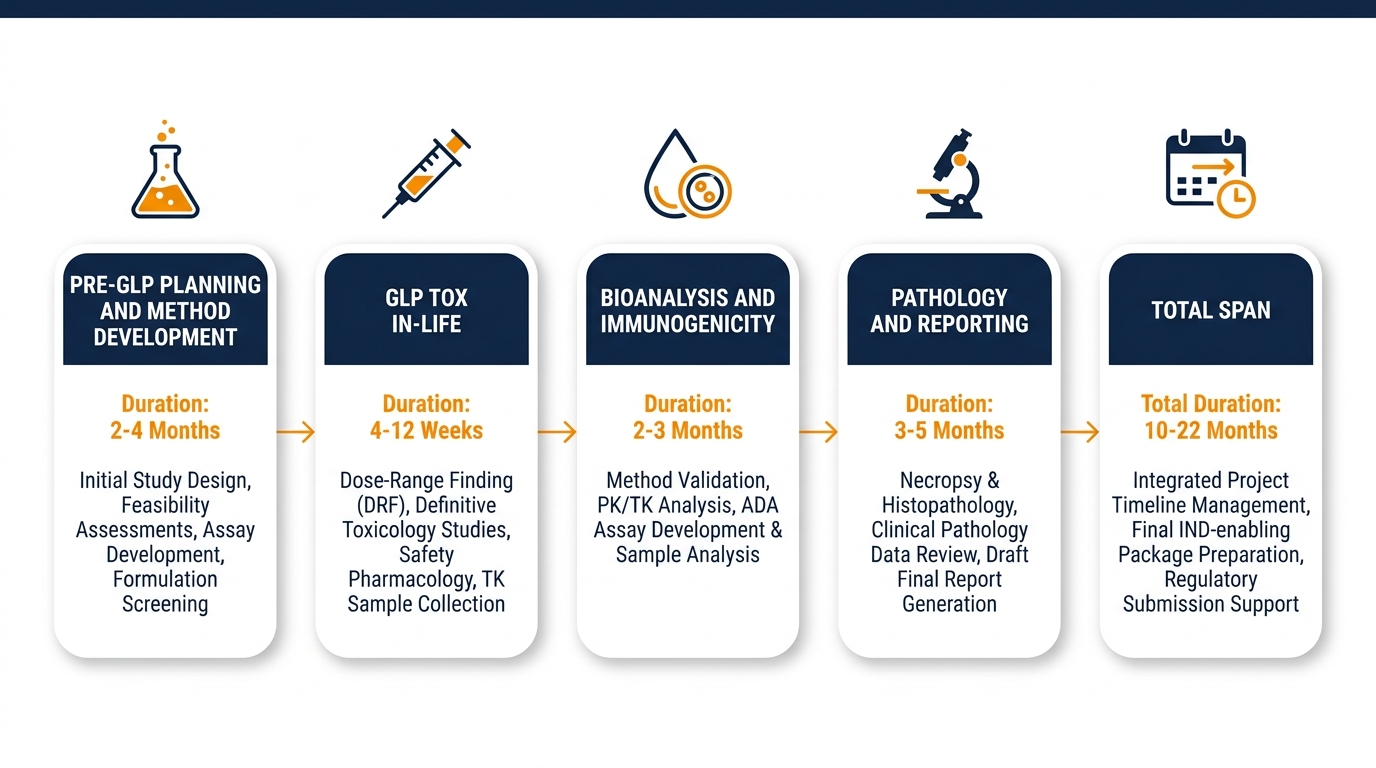
| Phase | Typical Duration | Key Activities |
|---|---|---|
| Pre-GLP planning & method development | 3–6 months | Bioanalytical assay development, model selection, pilot studies |
| GLP tox in-life | 3–9 months | Repeat-dose dosing, integrated safety pharmacology, TK sampling |
| Bioanalysis & immunogenicity | 2–4 months | Sample analysis, ADA assessment, data QC |
| Pathology & reporting | 3–6 months | Histopathology, peer review, GLP report finalization |
| Total span | 12–24 months | Variable by complexity and outsourcing strategy |
How Much Does a Preclinical Package for Biologics Cost?
Costs vary dramatically with modality and design, but full IND-enabling packages for biologics commonly run from several million to tens of millions of dollars. Major drivers include species choice (NHP studies are the most expensive), bioanalytical complexity, GLP compliance overhead, immunogenicity assay development, regulatory consulting, and CMC characterization.
Integrated programs that consolidate pharmacology, tox, PK/TK, and bioanalysis with one provider often reduce duplicated method transfers and timeline slippage — delivering both cost efficiency and scientific coherence.
Common Pitfalls When Building a Preclinical Package for Biologics
Critical Pitfalls to Avoid
- Inadequate test article characterization before GLP studies commence
- Selecting a convenient rather than pharmacologically relevant species
- Underestimating immunogenicity sampling needs and ADA interpretive framework
- Designing tox studies that do not mirror the planned clinical regimen
- Disconnects between preclinical endpoints and intended clinical biomarkers
- Delaying pre-IND regulatory consultation until after study completion
Choosing the Right CRO for Your Biologic Preclinical Package
Selection criteria should include demonstrated experience with the specific modality (mAb, ADC, fusion protein, gene therapy), regulatory track record, GLP-validated quality systems, transparent project management, and capacity for the relevant species. Communication cadence and scientific escort frequently determine whether issues are caught at the protocol stage or surface in the final report.
Consider partners with expertise in specialized capabilities such as large molecule bioanalysis / immunogenicity / PK, where assay design is tightly coupled to study interpretation. Facilities offering integrated large-animal capabilities, advanced surgical suites, and embedded scientific support shorten the path from protocol to defensible data.
| Sponsor Need | How an Integrated Preclinical Facility Helps |
|---|---|
| Single-site execution across pharmacology and tox | Consolidated study conduct reduces method transfers and timeline gaps |
| Relevant large-animal models | Access to swine, sheep, goat, and rabbit models with humanoid organ relevance |
| Scientific dialogue during design | Embedded scientific escort and brainstorming with senior investigators |
| Ethical conduct and 3R compliance | Documented animal welfare program and refinement-driven design |
| Transparent project management | Well-documented procedures and proactive sponsor communication |
| Tailored study designs | Flexible, science-based approach matching specific program needs and services |
Key Regulatory Guidelines for Biologic Preclinical Studies
The foundational documents include ICH S6(R1) for biotechnology-derived pharmaceutical safety evaluation, ICH M3(R2) for nonclinical study timing relative to clinical trials, ICH S7A and S7B for safety pharmacology, ICH Q5C for stability of biotechnological products, and ICH Q6B for specifications. Regional FDA and EMA guidances complement these for jurisdiction-specific expectations. Aligning study design with these references early reduces revision cycles during IND review.
ICH S6(R1)
Preclinical safety evaluation of biotechnology-derived pharmaceuticals — the primary guidance for biologic species selection and study design flexibility.
ICH M3(R2)
Nonclinical safety studies timing guidance — defines the scope and sequencing of studies relative to clinical trial phases.
ICH S7A / S7B
Safety pharmacology core battery and cardiac risk assessment — addresses integration of vital organ safety evaluation into biologic programs.
FAQ: Preclinical Package for Biologics
Ready to Plan Your IND-Enabling Program?
What does a confident IND submission look like for your specific biologic, modality, and clinical strategy? Our team brings more than 30 years of experience leading and managing large-animal preclinical research, with state-of-the-art facilities, embedded scientific escort, and a commitment to transparent collaboration.

This article is intended for scientific and informational purposes. All referenced ICH guidelines and regulatory frameworks are publicly available. For program-specific regulatory strategy, consult with qualified regulatory affairs professionals.




