IND-Enabling Toxicology: The Nonclinical Foundation Before First-in-Human
Before any drug candidate reaches human volunteers, a structured set of nonclinical safety studies must justify dosing. IND-enabling toxicology is that package — identifying target organ toxicities, establishing safety margins, and anchoring the first-in-human starting dose with defensible, regulator-ready data.
A well-structured IND-enabling program is not a checklist — it is a coherent scientific argument. Each study component must answer a specific regulatory question, and together they must tell a unified safety story that survives FDA and EMA scrutiny. Programs that integrate toxicokinetics, safety pharmacology, and repeat-dose toxicology from day one consistently reach FIH faster and with fewer amendments.
Which Toxicology Studies Are Considered Mandatory in Most IND Programs?
Most programs converge on a recognizable core: repeat-dose toxicity studies in two species (one rodent, one non-rodent), a safety pharmacology battery, and genotoxicity testing — all integrated with toxicokinetics to correlate exposure with effects. The exact scope is tailored to the molecule, indication, route of administration, and the planned duration of the Phase 1 trial.
Repeat-dose studies typically run 2–4 weeks for short-term FIH support. Safety pharmacology focuses on cardiovascular, central nervous system, and respiratory function. Genotoxicity is usually addressed via a three-test battery, while TK provides the exposure backbone. A well-designed GLP toxicology IND program is not a checklist — it is a coherent dataset where each study answers a specific regulatory and scientific question.
Repeat-Dose Toxicity
Two-species evaluation identifying target organs, NOAEL, and reversibility of effects across 2–4 week study durations.
Safety Pharmacology
Core battery covering cardiovascular, CNS, and respiratory function per ICH S7A/S7B, often integrated into repeat-dose protocols.
Genotoxicity Battery
Ames test, in vitro mammalian cell assay, and in vivo micronucleus assay — a short series with decisive go/no-go impact.
Is GLP Compliance Truly Required for IND-Enabling Toxicology?
For pivotal nonclinical studies intended to support human trials, compliance with Good Laboratory Practice (GLP) is generally expected by regulators. GLP is a quality system governing how nonclinical safety studies are planned, performed, monitored, recorded, archived, and reported. The result is a defensible, traceable dataset whose integrity can be independently verified by auditors and reviewers.
Early exploratory work — dose range finding (DRF), formulation screens, tolerability probes — is commonly performed non-GLP to conserve resources and refine dose selection. The definitive IND-enabling safety assessment, however, is run under GLP with an independent Quality Assurance unit. Foundational principles are codified in the OECD Principles on Good Laboratory Practice.
Practical Differences Between Non-GLP and GLP Toxicology
| Aspect | Non-GLP (Exploratory) | GLP (Pivotal) |
|---|---|---|
| Primary purpose | Internal decision-making, dose selection | Regulatory submission, FIH support |
| Protocol rigor | Flexible, iterative | Pre-approved, strictly followed |
| QA oversight | Minimal | Independent QA unit |
| Documentation | Lighter | Comprehensive, archived, traceable |
| Cost & timeline | Lower, faster | Higher, longer |
Realistic Timelines: How Long Does an IND-Enabling Safety Assessment Take?
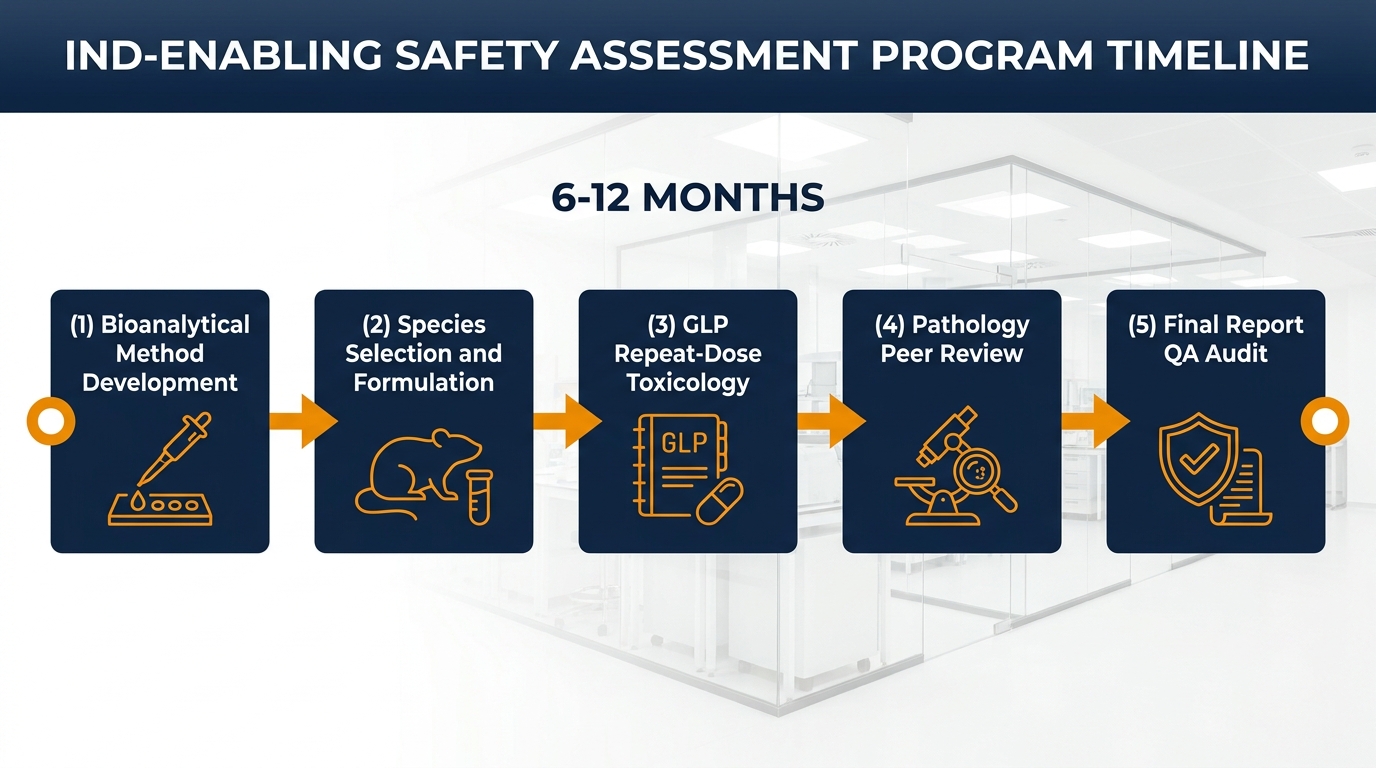
Programs commonly span 6–12 months from kickoff to IND-ready package, though specifics depend on test article supply, bioanalytical method development, repeat-dose duration, and CRO capacity. Bioanalytical validation for TK samples, pathology peer review, and final report QA auditing are recurring bottlenecks that shape the critical path.
Acceleration is achievable through smart design: integrating selected safety pharmacology endpoints into repeat-dose protocols, running genotoxicity in parallel with general toxicology, and front-loading risk assessment to avoid late surprises. The ICH M3(R2) guideline provides timing tables that anchor planning across regions.
- Integrate safety pharmacology endpoints into repeat-dose protocols where feasible
- Run genotoxicity studies in parallel with general toxicology — not sequentially
- Front-load bioanalytical method development before GLP studies commence
- Align formulation readiness with study start dates to prevent last-minute delays
Why the Repeat-Dose Toxicity Study Sits at the Center of IND
The repeat-dose toxicity study evaluates adverse effects when the test article is administered repeatedly across a defined period. It identifies target organs, characterizes the nature and reversibility of toxicity, and establishes the No-Observed-Adverse-Effect Level (NOAEL) — the anchor for human starting-dose calculations. Without it, exposure-effect interpretation collapses.
Standard endpoints include clinical observations, body weight and food consumption, hematology and clinical chemistry, urinalysis, organ weights, and gross pathology with histopathology of major organs. The study duration is matched to the planned human exposure: a 2–4 week study generally supports single-dose or short-term FIH protocols, while longer human dosing requires longer animal studies.
“The NOAEL is not just a number — it is a scientific argument. Its credibility depends on study design quality, exposure characterization, and the depth of pathological interpretation behind it.”
— Regulatory Toxicology Best Practice Principle
Deliverables You Should Receive From a Repeat-Dose Study
A complete repeat-dose package includes a full GLP study report, raw data audited by QA, pathology slides and blocks, integrated TK and bioanalytical data when applicable, and an expert toxicological interpretation tying findings to dose, exposure, and reversibility. These deliverables are what reviewers actually read — clarity and traceability matter as much as the science itself.
???? Complete Repeat-Dose Study Deliverables Checklist
- Full GLP-compliant study report with QA audit statement
- Archived raw data (paper and/or electronic) with full audit trail
- Histopathology slides and paraffin blocks for all target organs
- Integrated toxicokinetics and validated bioanalytical data
- Expert toxicologist interpretation: NOAEL, dose-response, reversibility assessment
Toxicokinetics (TK): Connecting Dose, Exposure, and Toxicity
Toxicokinetics measures systemic exposure (Cmax, AUC) of the test article and relevant metabolites within toxicology studies. Without TK, a finding at a given dose cannot be tied back to circulating drug levels, and safety margins relative to projected human exposure cannot be calculated with confidence. TK is what transforms a toxicology observation into a regulatory argument.
Robust, validated bioanalytical methods are prerequisites. Sample collection schedules must be designed so that key time points are captured without compromising the primary toxicology endpoints. The methodological expectations are described in detail in ICH S3A on toxicokinetics.
Design TK sampling schedules during protocol development — not as an afterthought. Pre-defined time points aligned with the PK profile of your molecule minimize satellite group requirements and reduce total animal use under 3R principles, while still capturing Cmax and AUC with statistical confidence.
Safety Pharmacology: Functional Liabilities You Cannot Skip
Safety pharmacology evaluates the potential adverse pharmacodynamic effects of a test article on vital organ systems at supratherapeutic doses. The core battery — defined under ICH S7A and complemented by S7B for QT-related risk — covers central nervous system function (e.g., functional observational battery), cardiovascular parameters (blood pressure, heart rate, ECG including QT), and respiratory measurements (rate, tidal volume).
Increasingly, sponsors integrate safety pharmacology endpoints into repeat-dose protocols when feasible. This shortens timelines, reduces animal use, and provides exposure-matched functional data. For programs requiring dedicated functional studies, the discipline is described in detail under Preclinical Safety Pharmacology Studies ICH S7A.
When Integration of Safety Pharmacology Into Repeat-Dose Studies Works
Integration is appropriate when measurements are non-invasive or minimally invasive, when the facility has validated instrumentation and trained staff, and when the added endpoints do not compromise primary toxicology readouts or impose undue stress on the animals. When these conditions are not met, a standalone study remains the safer choice scientifically and regulatorily.
CNS Assessment
Functional observational battery (FOB), motor activity, behavioral endpoints at supratherapeutic exposure.
Cardiovascular Safety
Blood pressure, heart rate, ECG analysis including QT interval — often via telemetry in non-rodent species.
Respiratory Function
Respiratory rate and tidal volume measurements; integrable into repeat-dose via whole-body plethysmography.
Genotoxicity Testing: A Short Battery With Outsized Impact
Genotoxicity testing assesses whether a test article can induce genetic damage — gene mutations, chromosomal aberrations, or aneuploidy. The standard battery includes a bacterial reverse mutation assay (Ames test), an in vitro mammalian cell assay (chromosome aberration or gene mutation), and an in vivo rodent micronucleus assay. A clean battery is a strong early signal supporting FIH progression.
These studies are often prioritized early in the IND-enabling timeline because they are relatively short and yield decisive go/no-go information. A positive finding can redirect chemistry, reframe risk, or stop a program — better discovered before pivotal repeat-dose investments. The framework is detailed in the ICH S2(R1) guideline.
A positive result triggers mechanistic follow-up to determine whether the signal is biologically relevant to humans. Depending on outcome, the program may continue with risk mitigation, pursue chemistry modifications, or be discontinued. Early genotoxicity testing is therefore a high-value de-risking investment — not a compliance formality.
Species Selection: Rodent, Non-Rodent, and the Biological Relevance Test
Regulatory expectations generally call for studies in two mammalian species — one rodent (typically rat) and one non-rodent (commonly dog, minipig, or non-human primate). Selection should be driven by biological relevance: similar metabolic profile, target expression, and pharmacological responsiveness. Practical factors such as model availability, route feasibility, and historical control data also matter.
For biotechnology-derived molecules, species selection often hinges on cross-reactivity with the human target. In some cases, a single pharmacologically relevant non-rodent species is scientifically justified — a position formally addressed in ICH S6(R1). Pig, rabbit, and sheep models are particularly informative for device-drug combinations and route-specific studies.
Default first species. Extensive historical control database, well-characterized metabolic pathways, cost-effective for repeat-dose studies.
Preferred for dermal, oral, and device-adjacent programs. Skin, GI tract, and cardiovascular anatomy closely parallel humans.
Highly relevant for cardiovascular, orthopedic, and lung device models. Surgical access and imaging feasibility are practical advantages.
Local Tolerance and Route-Specific Studies: When the Site Matters
When the route of administration could create unique local concerns not captured by systemic toxicology, dedicated local tolerance studies become necessary. Ocular formulations require assessment of irritation and ocular structure integrity. Dermal products demand evaluation of skin irritation, sensitization, and percutaneous absorption. Inhalation programs focus on respiratory histopathology and lung function.
Parenteral products (IV, IM, SC, intra-articular) require examination of injection-site reactions, inflammation, and tissue damage. These endpoints can be standalone studies or, in many cases, integrated into general toxicology protocols with additional histopathology and clinical observations at the administration site.
Reproductive Toxicology Before FIH: Required or Deferrable?
Reproductive and developmental toxicity studies (DART) are generally not required before FIH if women of childbearing potential are excluded and exposure is limited. This deferral, however, must be justified in the IND with a coherent clinical and contraceptive strategy. Deferral is a regulatory choice, not an oversight.
Earlier DART becomes necessary when women of childbearing potential are enrolled in early trials, when prolonged human exposure is anticipated, or when the mechanism of action raises specific reproductive concerns. The development plan, target population, and trial design jointly determine the right timing — best discussed with regulators during pre-IND interactions.
- Are women of childbearing potential excluded from Phase 1? → Deferral may be justified
- Does mechanism of action suggest reproductive risk? → Earlier DART warranted
- Is prolonged or repeat dosing planned in humans? → Discuss with regulators proactively
Comparison: IND-Enabling Toxicology vs Exploratory/DRF Studies
For a broader view of how these studies fit into the overall regulatory journey, see Biotech IND-enabling preclinical studies & filing.
| Dimension | Exploratory / DRF | IND-Enabling |
|---|---|---|
| Goal | Inform dose selection, internal decisions | Support IND submission and FIH |
| GLP status | Often non-GLP | GLP-compliant |
| Endpoints | Limited, focused | Comprehensive (clin path, histopath, TK) |
| QA oversight | Minimal | Independent QA audit |
| Output | Internal data summary | Defensible GLP report for regulators |
What the Pharmacology/Toxicology Section of an IND Must Contain
The nonclinical pharmacology/toxicology section is a structured narrative, not a data dump. It must summarize pharmacological activity (PK/PD in animals), primary and secondary pharmacology, the safety pharmacology battery, all toxicology studies (acute, repeat-dose, genotoxicity, specialty), and the integrated TK dataset. Each study description ties back to dose-response, exposure, and human relevance.
Critically, the section must integrate the data into a coherent safety assessment: NOAEL identification, exposure-based safety margin calculation, justification of the proposed starting dose, dose-escalation rationale, and a risk assessment with proposed clinical monitoring.
???? IND Pharmacology/Toxicology Section — Required Elements
- Pharmacological activity summary — PK/PD in animal models
- Safety pharmacology battery results (CNS, CV, respiratory)
- All toxicology study summaries with dose-response and NOAEL designation
- Integrated TK dataset with Cmax and AUC data by species
- Human starting dose justification with exposure-based safety margin calculation
Common Mistakes That Delay or Derail an IND-Enabling Program
Three patterns recur. First, underinvesting in bioanalytical method development, which then becomes the rate-limiting step for TK and final reporting. Second, selecting species for convenience rather than biological relevance, leading to reviewer questions and potential bridging studies. Third, treating safety pharmacology as an afterthought — discovering a QT signal late in development is among the most expensive findings in the industry.
A fourth, less obvious mistake is poor coordination between toxicology, CMC, and clinical teams. The toxicology dose range must be achievable with the available formulation; the route must be clinically realistic; the duration must align with the intended Phase 1 protocol. Misalignment forces repeat work, not just paperwork.
Method development left too late becomes the critical-path bottleneck for TK data and final GLP report completion.
Choosing species based on availability rather than target relevance generates reviewer questions and may require costly bridging studies.
Safety pharmacology deferred to late development — a QT liability found after significant investment is one of the industry’s costliest findings.
Choosing a CRO for IND-Enabling Toxicology Studies

CRO selection is a risk decision. Evaluate regulatory acumen with FDA and EMA submissions, scientific depth across toxicology, pathology, and bioanalysis, demonstrable GLP compliance with an independent QA unit, facility capacity matched to your timeline, and communication discipline. A partner that surfaces problems early is worth more than one that reports only successes.
| Business Need | What a Capable Partner Provides |
|---|---|
| Strategic nonclinical plan | Gap analysis tied to target indication and clinical design |
| Species relevance for biologics or devices | Access to large-animal models (pig, sheep, rabbit) with surgical capability |
| Route-specific or combination product testing | Imaging-equipped surgical suites and integrated study design |
| Regulatory defensibility | GLP-compliant execution with full documentation and archiving |
| Timeline compression | Integrated safety pharmacology + repeat-dose + TK in one protocol |
| Sponsor visibility | Transparent collaboration, scientific escort, clear reporting cadence |
How Biotech Farm Supports IND-Enabling Toxicology Programs
Biotech Farm operates as a preclinical R&D facility focused on large-animal models and complex study designs that smaller laboratories cannot accommodate. Programs benefit from tailored study design and strategic consulting, GLP-aligned repeat-dose toxicology in biologically relevant species, comprehensive safety pharmacology coverage (cardiovascular, CNS, respiratory), genotoxicity coordination, and integrated toxicokinetics with bioanalytical support.
The relative advantage is practical: state-of-the-art surgical suites with C-Arm fluoroscopy, high-definition ultrasound, echocardiography, and 4K laparoscopic capability allow route-specific and device-drug combination studies under one roof. The animal facility is operated with documented procedures, ethical oversight aligned with the 3Rs (Replacement, Reduction, Refinement), and a professional team with decades of research management experience.
C-Arm fluoroscopy, HD ultrasound, echocardiography, and 4K laparoscopic systems enabling route-specific and device-drug combination studies.
Pig, sheep, rabbit, and dog models with documented animal care procedures and full ethics committee oversight aligned with 3R principles.
From protocol to final GLP report — toxicology, safety pharmacology, TK, and bioanalysis coordinated under one scientific leadership structure.
What Distinguishes Biotech Farm as a Nonclinical Development Partner
Sponsors gain access to experienced toxicologists and senior surgeons, customizable study designs that match the molecule rather than a fixed template, and integrated execution from protocol to final report. Transparent collaboration and proactive scientific escort reduce the friction that typically appears between sponsor and CRO during pivotal studies — a difference most visible during regulatory questions and amendments.
“A partner that surfaces problems early is worth more than one that reports only successes. Regulatory defensibility is built study by study — and it starts with the right scientific and operational foundation.”
— Biotech Farm Preclinical Development Philosophy
Frequently Asked Questions on IND-Enabling Toxicology
Ready to Plan Your IND-Enabling Toxicology Program?
What does your nonclinical safety package look like today, and where are the gaps that could delay your IND? If you would like a structured discussion on study design, species selection, GLP execution, or regulatory strategy, the team at Biotech Farm is available to review your program and propose a tailored path forward.





