Expert Analysis — IND-Enabling Study Design
IND-Enabling Study Design: A Structured Path From Discovery to First-in-Human
Drawing on over 20 years of large-animal model expertise and more than a decade of collaborative veterinary research, the BIOTECH FARM team delivers scientifically composed, ethics-driven IND-enabling packages — from protocol design through GLP-aligned execution — for both industry and academic partners worldwide.
⚡ Expert Insight
IND-enabling study design is not simply a checklist of required studies — it is the strategic architecture that determines whether a molecule transitions safely and efficiently into human trials. Programs that align formulation, species selection, dose justification, and GLP execution from day one consistently outperform those that treat these elements as independent tasks.
What Is IND-Enabling Study Design?
IND-enabling study design is the systematic planning of a nonclinical research package intended to support an Investigational New Drug (IND) application. The objective is to generate sufficient safety, exposure, and activity evidence so that regulators can decide whether human clinical trials may proceed.
The design process defines regulatory questions, selects appropriate models, determines endpoints, dosages, and administration regimens, and ensures data quality — including Good Laboratory Practice (GLP) where required. Beyond running studies, it also plans for the integration of the resulting data into the submission documents themselves.
In practice, IND-enabling design is the critical bridge between early drug discovery and first-in-human (FIH) clinical trials, and the quality of that bridge directly affects timelines, regulatory feedback, and the safety of the volunteers who will eventually receive the drug.
Are IND-Enabling Studies the Same as “Preclinical” or “Nonclinical” Work?
Although the three terms are often used interchangeably, they describe different scopes. Preclinical is the broadest umbrella, covering all research conducted prior to human clinical trials, including early discovery, in vitro assays, and efficacy models. Nonclinical emphasizes studies not conducted in humans that are performed for regulatory purposes — toxicology, pharmacokinetics (PK), safety pharmacology, and ADME. IND-enabling is a focused subset of nonclinical work, tailored specifically to address the regulatory expectations for initiating human trials.
Why This Distinction Matters in Planning
Confusing the three categories often leads to over-spending on early studies that cannot support an IND, or under-investing in the GLP-grade pivotal package regulators expect. Clear scoping at the start prevents both extremes.
| Dimension | Preclinical | Nonclinical | IND-Enabling |
|---|---|---|---|
| Scope | All pre-human research | Non-human studies for regulatory use | Subset directly supporting IND |
| Typical Purpose | Discovery, PoC, efficacy | Tox, PK, safety pharmacology, ADME | FIH safety, dose, monitoring |
| GLP Expected? | Often not | For pivotal safety studies | Yes, for pivotal package |
| Output | Hypothesis support | Regulatory dossier modules | IND submission readiness |
Which Nonclinical Studies Are Typically Required to Support an IND?
The exact package depends on the drug product, indication, and planned clinical duration, but most programs include pharmacology (mechanism of action), ADME/PK, repeat-dose toxicology, safety pharmacology core battery, genotoxicity, and — when relevant — immunotoxicity and reproductive toxicology.
Requirements follow a risk-based logic that considers route of administration, duration of treatment, patient population, novelty of the modality, impurities and excipients, and systemic exposure levels. The ICH M3(R2) guideline provides the core framework.
Core Safety Studies
- Repeat-dose toxicology
- Safety pharmacology (ICH S7A/S7B)
- Genotoxicity battery
PK/ADME Studies
- Absorption, distribution, metabolism, excretion
- Toxicokinetics (integrated with tox studies)
- Drug-drug interaction screens
Conditional Studies
- Reproductive / developmental toxicology
- Immunotoxicity
- Carcinogenicity (indication-dependent)
Building a Nonclinical Study Plan: A Step-by-Step Approach

A robust IND-enabling plan is iterative rather than linear. It typically begins with defining the Target Product Profile (TPP) and the clinical use scenario, so every study has a clear translational purpose.
Establish the clinical use scenario so every study has a clear translational purpose and regulatory rationale.
Identify gaps and determine which questions are already answered by existing nonclinical evidence.
Range-finding before pivotal toxicology; in vitro screens before in vivo PK — each milestone gates the next investment.
Summary tables and a coherent safety narrative tie nonclinical data to clinical assumptions.
Prioritize mandatory pre-IND milestones versus those that may follow initial submission.
???? Case Study
Small Biotech: Aligning Protocol and Pivotal Toxicology
A small molecule program targeting a chronic indication with an intended 6-month clinical regimen must commit early to dosing route, formulation, and a pharmacologically relevant species. If the chosen toxicology formulation diverges from the clinical formulation later, bridging studies become inevitable, and the timeline expands. A well-aligned plan locks in formulation and route before the pivotal repeat-dose toxicology study begins, ensuring that the eventual NOAEL and exposure margins translate cleanly to the proposed clinical dose. This kind of upfront alignment is one of the most common differentiators between programs that move smoothly through pre-IND interactions and those that face holds.
What Should an IND-Enabling Nonclinical Protocol Contain?
A defensible nonclinical protocol clearly states study objectives (e.g., determine NOAEL, characterize PK), the rationale linking it to overall IND strategy, and the experimental design — species, strain, group sizes, dose levels, route, duration, and follow-up.
Study Objectives & Rationale
Clear statement of what the study is designed to determine and why it is needed for the IND.
Experimental Design
Species, strain, group sizes, dose levels, route, duration, recovery groups, and follow-up periods.
Endpoints & Sampling
Clinical observations, body weight, food consumption, clinical pathology, histopathology, PK, and biomarker schedules.
Methods & Analytics
Animal care, drug preparation, sample processing, bioanalysis, necropsy, and bioanalytical method validation status.
Statistical Plan
Data acceptability criteria, stopping rules, and pre-specified statistical methods for all primary endpoints.
GLP Compliance & QA
GLP status declaration, planned QA audits, archiving procedures, and deviation management protocol.
Choosing Species and Models for IND-Enabling Toxicology
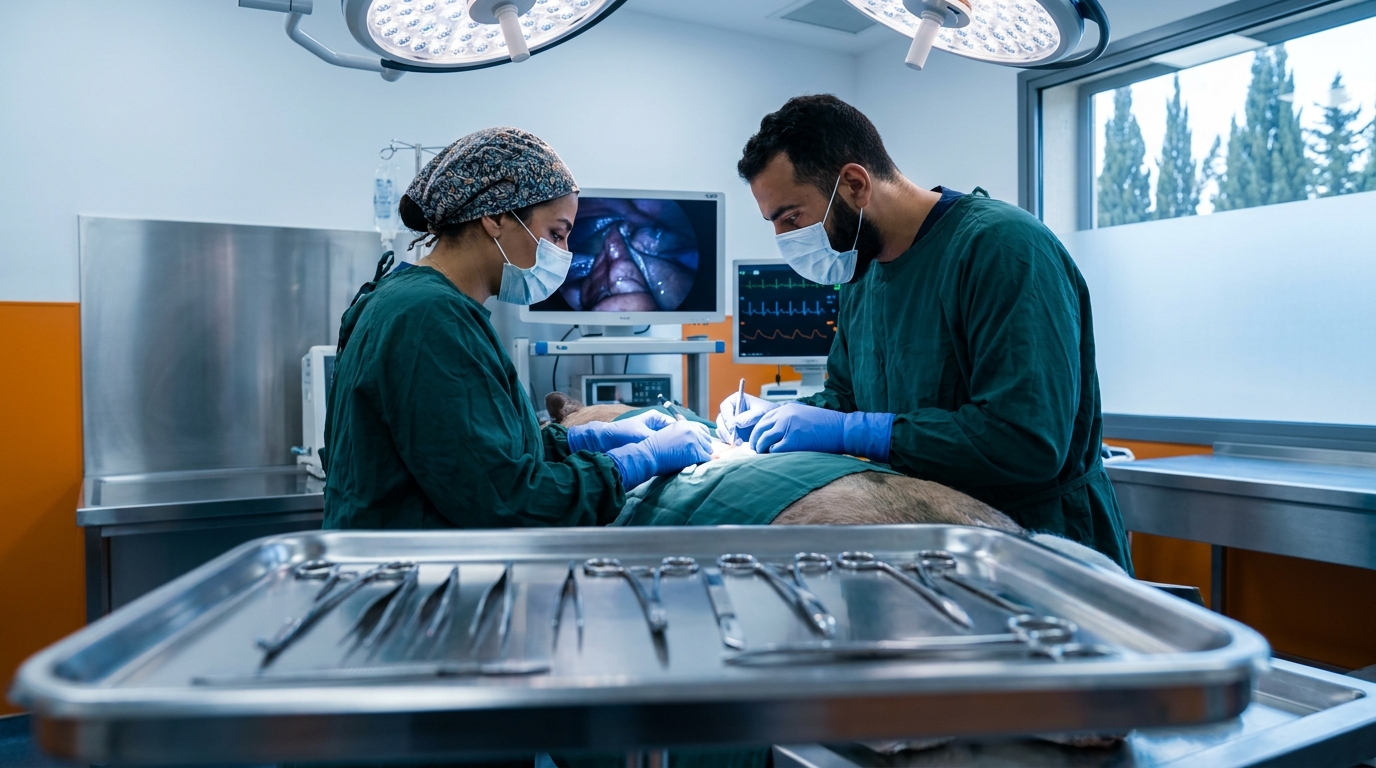
Species selection is one of the most consequential decisions in the entire program. The chosen species should show similar target binding and pharmacological response to humans, metabolize the drug in a comparable way, tolerate the intended clinical route, and be physiologically relevant to the toxicities of concern.
The ICH S6(R1) guideline is especially important for biotechnology-derived products, where species relevance often dominates the discussion. A second species may not add value when only one species is pharmacologically relevant or when the second yields no new findings — but the justification must be scientifically robust.
???? Large-Animal Expertise
BIOTECH FARM’s large-animal platforms — pig, sheep, and rabbit — offer humanoid-organ relevance for cardiology, ophthalmology, orthopedics, and respiratory indications where rodent models fail to replicate human anatomy and physiology. On-site C-Arm fluoroscopy, echocardiography, OCT, and 4K laparoscopic capabilities enable imaging-driven endpoints that translate directly into the clinical monitoring strategy.
Selecting Dose Levels and Escalation for Repeat-Dose Toxicology
Dose selection in pivotal repeat-dose studies is designed to bracket the safety profile: a low dose with minimal or no toxicity, a high dose that produces toxicity (or is limited by formulation, solubility, or maximum feasible volume), and a mid dose that informs the dose-response curve.
Range-finding studies establish the working window, and preliminary PK confirms adequate systemic exposure at each level. Exposure multiples (AUC, Cmax) relative to anticipated human exposure justify the safety margin. Predefined stopping criteria protect animal welfare and scientific integrity.
“ICH M3(R2) provides guidance on appropriate exposure margins and the duration of repeat-dose studies needed to support each clinical phase — from single-dose FIH studies through long-term chronic treatment.” — ICH M3(R2) Core Framework
What Is GLP, and Which IND-Enabling Studies Must Comply?
Good Laboratory Practice (GLP) is the quality system governing how nonclinical safety studies are planned, performed, monitored, recorded, archived, and reported. It ensures the integrity and reliability of data submitted to regulators. The OECD Principles of GLP describe the international framework, and 21 CFR Part 58 codifies the U.S. requirements.
✅ GLP Required
- Pivotal repeat-dose toxicology
- Safety pharmacology core battery
- Genotoxicity studies
- Reproductive toxicity (where applicable)
⚠️ GLP Not Required (Typically)
- Target identification studies
- Early lead screening
- Range-finding dose studies
- Exploratory PK/PD studies
How Does Safety Pharmacology Fit Into IND-Enabling Design?
Safety pharmacology investigates undesirable pharmacodynamic effects on vital organ systems at and above therapeutic exposures. The ICH S7A core battery covers the cardiovascular, central nervous, and respiratory systems, and ICH S7B addresses the QT prolongation risk specifically.
Findings from these studies shape clinical monitoring strategies, exclusion criteria, and the proposed escalation scheme for FIH. Some endpoints — telemetered ECG, for example — can be integrated into repeat-dose toxicology when conditions allow, which can compress timelines without compromising regulatory acceptability.
???? Integration Opportunity
Programs that build a coordinated preclinical safety pharmacology approach tend to surface risk earlier and avoid late-stage redesigns. Integrating ECG, blood pressure, and respiratory endpoints into repeat-dose studies — where scientifically appropriate — reduces animal use, compresses timelines, and generates richer data from fewer animals.
What Does ADME/PK Drive in an IND-Enabling Program?
ADME/PK studies quantify how the body absorbs, distributes, metabolizes, and excretes the drug, producing parameters such as Cmax, Tmax, AUC, and half-life. These data underpin exposure-response interpretation, dose selection across species, metabolite characterization, drug-drug interaction risk assessment, and clinical monitoring strategy.
Bioanalytical methods must be developed and validated against regulatory expectations before they support GLP toxicokinetic samples; otherwise the entire safety dataset becomes vulnerable to regulator questions. PK sampling schedules should be designed to capture absorption, distribution, and elimination phases reliably — not merely convenient time points.
Translating NOAEL Into a Safe First-in-Human Starting Dose
Translating the No Observed Adverse Effect Level (NOAEL) into an FIH starting dose follows a defined logic: identify the NOAEL in the most relevant species (by mg/kg or exposure), convert to a Human Equivalent Dose (HED) using species-appropriate scaling, apply a safety factor (commonly ≥10) reflecting inter- and intra-species variability, severity of findings, dose-response steepness, modality novelty, and overall data quality.
For highly potent drugs and many biologics, the Minimum Anticipated Biological Effect Level (MABEL) may govern starting dose instead of, or together with, NOAEL. PK/PD modeling refines the prediction. The FDA guidance on maximum safe starting dose describes the algorithm in detail.
No Observed Adverse Effect Level — highest dose without adverse findings in nonclinical studies.
Human Equivalent Dose — species-scaled conversion using body surface area or allometric methods.
Minimum Anticipated Biological Effect Level — governs starting dose for highly potent drugs and certain biologics.
Common IND-Enabling Design Mistakes That Trigger Regulator Questions
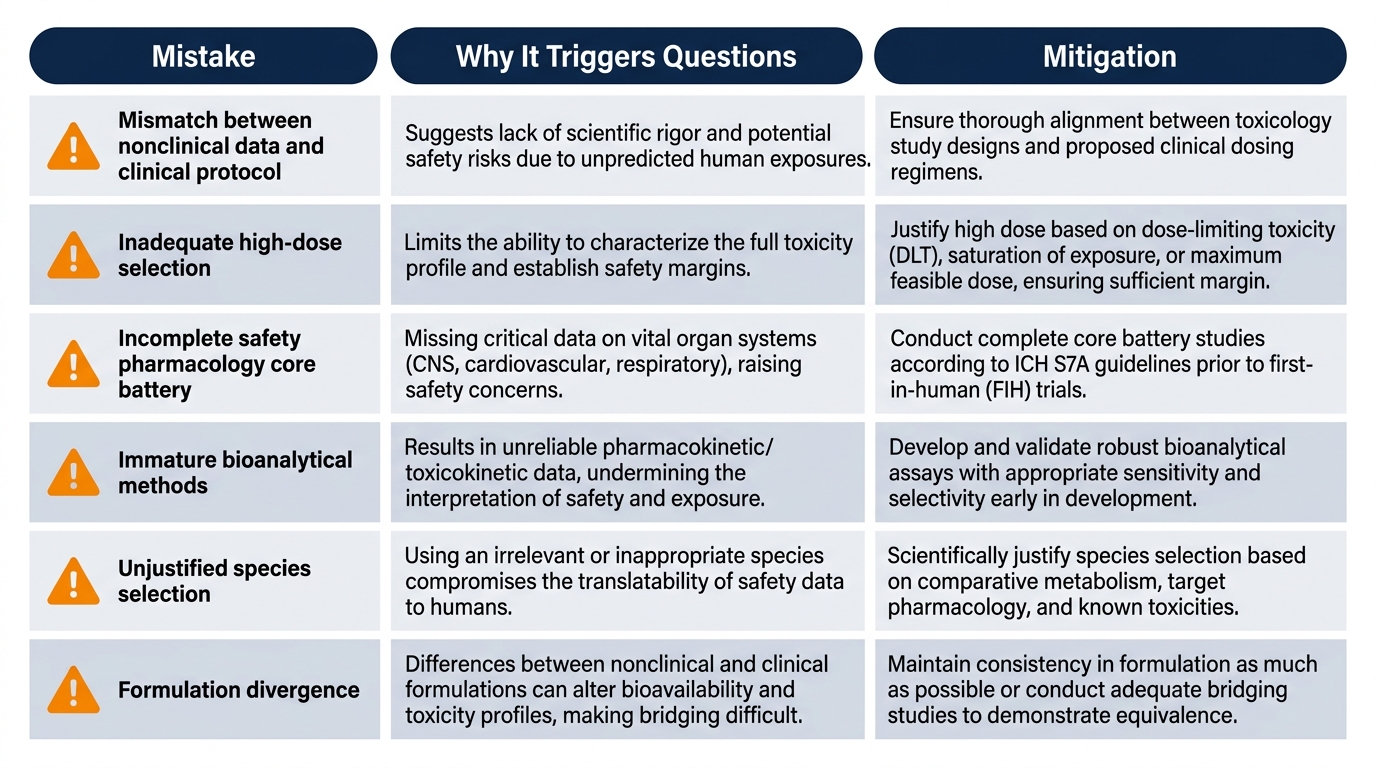
| Mistake | Why It Triggers Questions | Mitigation |
|---|---|---|
| Mismatch between nonclinical data and clinical protocol | Insufficient exposure margins at proposed clinical dose | Lock TPP and clinical scenario before pivotal tox |
| Inadequate high-dose selection | No NOAEL identifiable; safety margins unjustified | Use range-finding plus PK to confirm exposure |
| Incomplete safety pharmacology core battery | Cardiovascular/CNS/respiratory risks unaddressed | Plan ICH S7A endpoints early, integrate where feasible |
| Immature bioanalytical methods | Unreliable toxicokinetic data | Validate before GLP samples are generated |
| Unjustified species selection | Pharmacological relevance unclear | Document target homology, metabolism, and response |
| Formulation divergence (tox vs. clinical) | Bridging data missing | Align formulations or pre-plan bridging studies |
⚠️ High-Risk Warning
Bioanalytical methods that are not fully validated before GLP toxicokinetic samples are generated put the entire safety dataset at risk. This is among the most frequent — and most preventable — sources of regulatory questions in IND submissions.
How Long Do IND-Enabling Studies Realistically Take?
Timelines vary with modality, complexity, planned clinical duration, CRO capacity, and regulatory feedback. A small molecule program typically requires 12–18 months from the start of pivotal toxicology to IND submission, while biologics often extend to 18–24 months or more due to longer studies in relevant species.
| Activity | Typical Duration |
|---|---|
| Protocol finalization and animal sourcing | 1–2 months |
| Repeat-dose toxicology with histopathology | 3–6 months |
| Safety pharmacology core battery | 1–3 months |
| Genotoxicity battery | 1–2 months |
| Bioanalytical development and validation | 1–3 months |
| Report writing (per major study) | 2–4 months |
| Pre-IND meeting preparation and IND assembly | 3–6 months |
Compression is possible, but never at the expense of data quality. Programs structured as integrated IND-enabling preclinical packages tend to compress sensibly because studies share infrastructure, animals, and bioanalytical capacity.
How a Large-Animal IND-Enabling Partner Adds Value
| Program Need | How an Integrated Facility Supports It |
|---|---|
| Translatable models | Large-animal platforms (pig, sheep, rabbit) with humanoid-organ relevance for cardiology, ophthalmology, orthopedics, and respiratory work |
| Imaging-driven endpoints | On-site C-Arm fluoroscopy, echocardiography, OCT, and 4K laparoscopic capabilities for in-life functional assessment |
| Scientific escort | Protocol design, endpoint selection, and bioanalytical planning supported across the program rather than per study |
| GLP-aligned execution | Documented procedures, QA practices, and data integrity aligned with regulatory expectations for pivotal safety work |
| Ethical operation | 3R principles (Replacement, Reduction, Refinement) embedded in animal welfare and study planning |
“The most impactful contribution a large-animal research partner makes is not simply running a study — it is designing studies that answer regulatory questions with the first experiment, not the third.” — Adir Koreh, CEO, BIOTECH FARM Ltd.
Frequently Asked Questions on IND-Enabling Study Design
Planning Your IND-Enabling Program?
Which part of your nonclinical package still needs alignment between scientific design and regulatory expectation — species selection, dose justification, or integration of safety pharmacology with repeat-dose toxicology? The team at BIOTECH FARM can help you translate strategy into a concrete, defensible plan.





